

 PDF
PDF Citation
Citation Print
Print


Introduction
Vascular endothelial cells have important physiologic functions in maintaining cardiovascular stability. Many risk factors can cause endothelial cell dysfunction and result in apoptosis. The production of reactive oxygen species (ROS) is considered a crucial pathogenic factor in endothelial cell dysfunction and the development of cardiovascular diseases such as hypertension, atherosclerosis, and diabetic vasculopathy. Hydrogen peroxide (H2O2), a major ROS, plays a crucial role in vascular dysfunction [1]. Many studies have revealed that H2O2 can damage and induce apoptosis in endothelial cells, the underlying mechanisms of which are associated with mitogen activated protein kinases (MAPKs) and Akt [23]. Among the MAPK members, cJun-N-terminal kinases (JNK) and p38 MAPK are preferentially activated in response to various stresses and pro-apoptotic signals in numerous cell types. The Akt pathway is commonly linked to survival pathways against multiple pro-apoptotic stimuli that induce its activation [4]. Thus, blockade of these pro-apoptotic pathways in endothelial cells is regarded as a potential therapeutic strategy to prevent or alleviate the progression of H2O2-induced cardiovascular diseases.
Propofol (2,6-diisopropylphenol) is an intravenous sedative/hypnotic agent widely used for anesthesia and sedation. Its structure is similar to that of the endogenous antioxidant α-tocopherol (Vitamin E) [5]. Propofol has anti-oxidant and anti-apoptotic effects [2678], but these vary among cell types and involve multiple mechanisms. Propofol differentially activates certain MAPKs depending on the cell type used and on the experimental conditions [2910]. Additionally, the role of Akt in propofol-mediated anti-apoptotic processes remains controversial. Propofol has been shown to protect cardiac H9c2 cells against H2O2-induced injury by activating Akt [8]. However, Akt is not activated by H2O2 treatment, nor is it regulated by propofol pretreatment in human umbilical endothelial cells (HUVECs) [3]. Although the anti-apoptotic effect of propofol on vascular endothelial cells has been investigated extensively, the underlying mechanisms remain unclear.
This study aimed to examine the anti-apoptotic effects of propofol on HUVECs and evaluate the potential mechanisms. Our results suggested that the protective effect of propofol against H2O2-induced HUVEC dysfunction was most likely due to its anti-apoptosis action. Our study also demonstrated the involvement of the p38 MAPK, JNK and Akt pathways in this process.
Materials and Methods
Reagents
Propofol (purity >97%; Lot number: D126608) was obtained from Sigma Aldrich (St. Louis, USA). A Cell Counting Kit-8 (CCK-8) was purchased from Dojindo laboratories (Kumamoto, Japan). A bicinchoninic acid (BCA) Protein Assay Kit was obtained from the Beyotime Institute of Biotechnology (Haimen, China). The Colorimetric CaspACE™ Assay System was purchased from Promega (Madison, WI, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and rabbit polyclonal antibodies for Akt, phosphorylated (p)-Akt (Ser473), p38MAPK, phosphorylated (p)-p38MAPK, JNK, phosphorylated (p)-JNK, Bax, Bcl-2, and β-actin were obtained from Cell Signaling Technology (Beverly, MA, USA). Dimethyl sulfoxide (DMSO) was purchased from Biosharp (Hefei, China). All other chemicals and reagents used were commercially available and of standard biochemical quality.
Cell culture and treatment
HUVECs were obtained from human umbilical cord veins by digestion with 0.1% collagenase type II (Sigma) as previously described [11]. The cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Hyclone, UT, USA) supplemented with heat-inactivated 15% fetal bovine serum, endothelial cell growth supplement, 100 U/ml penicillin, and 100 µg/ml streptomycin. The cells were then incubated at 37℃ in 5% CO2 and 95% air. HUVECs at passages 2-5 were used for the experiments. Blood vessels were labeled with anti-von Willebrand factor antibody [12]. After 1 week of culture, HUVECs were identified via immunofluorescence staining with an anti-von Willebrand factor antibody [13].
HUVECs were cultured and divided into eight groups depending on the treatment: Group C1 without any treatment was used as the control; cells in groups C2, P, and H were incubated with 0.1% DMSO, 50 µM propofol, and 400 µM H2O2, respectively, for 4 h. In groups P1+H, P5+H, P25+H and P50+H, cells were treated with 1, 5, 25, and 50 µM propofol, respectively, for 30 min and then incubated with 400 µM H2O2 for 4 h. Propofol was prepared in DMSO and diluted with culture medium immediately before use.
Cell viability assay
Cell viability was tested using the CCK-8, according to the manufacturer's instructions [14]. Cells (2 × 103 /well) were seeded in a 96-well flat-bottomed plate and treated with the indicated conditions at 37℃. After 24 h, 10 µl of WST-8 dye were added into each well, the cells were incubated at 37℃ for 1 h, then the optical density at 450 nm was measured using a microplate absorbance reader Elx800 (Bio-Tek, Instruments, Inc., Winooski, VT, USA). Cells that stained positively with the CCK-8 solution were considered viable and are presented as percentages compared with control cells.
Annexin V-fluorescein isothiocyanate/propidium iodide (AV-FITC/PI) double-staining assay
HUVECs treated with propofol and/or H2O2 were collected via trypsin digestion, washed with cold phosphate-buffered saline (PBS), and resuspended in Annexin V binding buffer. AV-FITC [Becton Dickinson (BD) Biosciences, USA] at a final concentration of 1 µg/ml and 250 ng of PI were added to a mixture containing 100 µl each of cell resuspension and binding buffer (BD Biosciences, USA). The mixture was incubated in the dark for 15 min at room temperature. Cells were washed once with binding buffer and resuspended in 400 µl of binding buffer prior to flow cytometric analysis using a BD FACScan Flow Cytometer (BD, Mountain View, USA).
Caspase-3 activity assay
HUVECs were lysed and caspase-3 activity was measured in the cell lysate using the CaspACE™ Assay System. The colorimetric caspase-3-specific substrate, N-acetyl-Asp-Glu-Val-Asp-p-nitroaniline that was provided within the Assay System was labeled with the chromophore p-nitroaniline (pNA). Free pNA produces a yellow color that is monitored by spectrophotometry at 405 nm. The degree of yellow color produced upon cleavage was proportionate to the DEVDase (caspase-3) activity present in the sample.
Western blotting
Cells in a six-well plate were washed with PBS and lysed with lysis buffer containing phenylmethanesulfonyl fluoride (PMSF) at 4℃ for 5 min. The lysates were collected and centrifuged at 14,000 rpm and 4℃ for 15 min, and the supernatant was collected for protein analysis using an Enhanced BCA Protein Assay Kit. The whole protein samples were boiled for 15 min in 5×loading buffer and separated using 10% sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE). Samples (50 µg/lane) were loaded on 10% SDS-PAGE, and proteins were transferred to polyvinylidene fluoride membranes at a current of 300 mA for 90 min. The membranes were incubated in Tris-buffered saline containing 0.1% Tween-20 and 5% dry milk, and then incubated overnight with anti-GAPDH, β-actin, Bax, Bcl-2, caspase-3, p38MAPK, p-p38MAPK, JNK, p-JNK, Akt or p-Akt (Ser473) antibodies. After 2 h of incubation with anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies, the protein bands were detected using an enhanced chemiluminescence (ECL) kit. The immune complexes were visualized via fluorography using an enhanced ECL system (Millipore, USA).
Statistical analysis
Data analysis was performed using GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA). All experimental data were measured at least three times. The data were expressed as means ± SD, and results were analyzed by one-way ANOVA. A value of P <0.05 was taken to indicate statistical significance.
Results
Propofol pretreatment attenuated HUVEC cytotoxicity induced by H2O2
Cell viability was measured using the CCK-8 kit. After incubation with 100, 200, 300, 400 and 500 µM H2O2 for 4 h, the viability of HUVECs was reduced significantly, in a dose-dependent manner (Fig. 1). When cells were treated with 400 µM H2O2 for 4 h, the cell viability was ~50% of the control. Therefore, 400 µM H2O2 was used in subsequent experiments. Propofol alone did not affect cell viability as significantly as in the control group; however, pretreatment with propofol inhibited the H2O2-induced cytotoxicity in a dose-dependent manner (Fig. 2).
Propofol reduced HUVEC apoptosis and necrosis induced by H2O2
Using Annexin V/PI staining, we examined the protective role of propofol in H2O2-induced apoptosis. The apoptosis rate was the sum of early and late apoptosis events. As shown in Fig. 3, after 4 h of treatment with 400 µM H2O2, the proportion of apoptotic/necrotic cells was more than threefold that of the untreated control (Fig. 3B). Pretreatment with propofol significantly decreased H2O2-induced apoptosis. At 5, 25 and 50 µM, propofol markedly decreased the percentage of apoptotic cells following H2O2 treatment.
Propofol decreased caspase-3 activity in HUVECs induced by H2O2
H2O2 led to a significant increase in caspase-3 activity. At 25 and 50 µM, propofol significantly reduced H2O2-induced caspase-3 activation (Fig. 4A).
Effects of propofol on Bcl-2, Bax, and caspase-3 protein levels in HUVECs treated with H2O2
Bax expression was increased and that of Bcl-2 was decreased following H2O2 treatment. However, both changes were suppressed by pretreatment of cells with propofol (Figs. 4B and 4C).
The expression levels of caspase-3 in HUVECs were increased following H2O2 treatment. These increases were markedly reduced in the propofol-pretreated groups, in a dose-dependent manner (Figs. 4B and 4D).
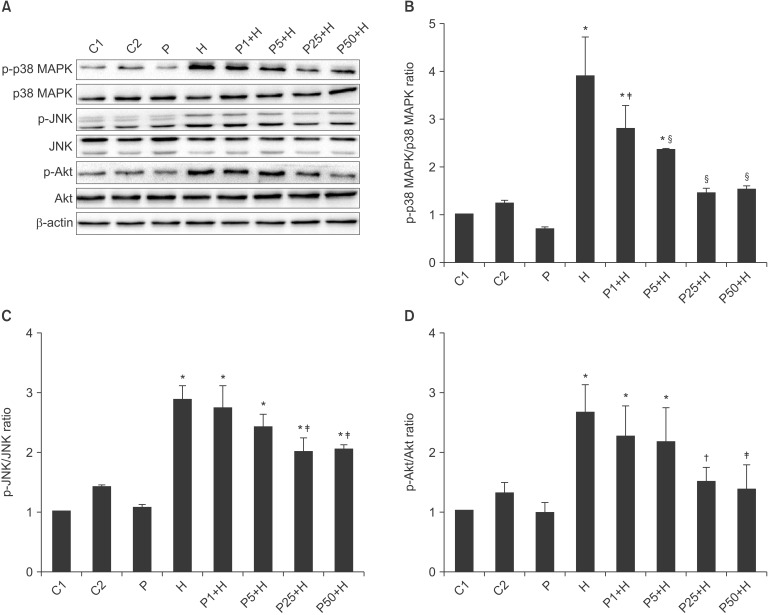
Effects of propofol on p38 MAPK/JNK/Akt phosphorylation
To determine the mechanisms of action of propofol, p38 MAPK, p-p38 MAPK, JNK p-JNK, Akt, and p-Akt (Ser473) were detected using western blotting. The results showed that H2O2 significantly activated the p38 MAPK, JNK and Akt signaling pathways. Pretreatment of cells with propofol attenuated this effect of H2O2 (Fig. 5).
Discussion
Propofol has been shown to have antioxidant and antiapoptotic effects [15]. In this study we showed that propofol protected HUVECs from cytotoxic reactive oxygen species in a dose-dependent manner. We found that the minimum effective concentration of propofol was 1 µM, which was equal to the initial concentration for sedation [16]. Since steady-plasma concentrations of propofol in a clinical setting are 10 to 50 µM [17], we examined the effect of propofol at 1-50 µM. In human leukocytes, the antioxidant effect of propofol is due in part to intralipid, the solvent used to deliver propofol to patients [18]. Accordingly, we chose dimethyl sulfoxide (DMSO) as the solvent to deliver propofol to the cells. However, DMSO has recently been proposed as a free radical scavenging agent [19]. To exclude the potential effect of DMSO, we included a DMSO control group in our experiments. The results showed that DMSO had no effect on H2O2-induced changes. Therefore, we concluded that propofol, rather than the solvent, affected the HUVECs.
The members of the Bcl-2 family of proteins serve as anti- or pro-apoptotic regulators that are associated with a wide variety of cellular activities [420]. Bcl-2 is an anti-apoptotic regulator and Bax is a pro-apoptotic regulator. The balance between anti- and pro-apoptotic proteins partly determines the fate of the cell. On the other hand, caspases are a family of specific cysteine proteases whose activation is crucial to intracellular apoptosis. Among several identified caspases, caspase-3 is a common target of several procaspases and caspases, and therefore, can act as an apoptotic marker induced by various stimuli [4]. It has been shown that H2O2-induced apoptosis in cardiomyocytes is ameliorated by propofol through up-regulation of Bcl-2 and inhibition of caspase-3 [8]. In the present study, H2O2 resulted in a reduced Bcl-2/Bax ratio in HUVECs. At 1 to 50 µM, pretreatment with propofol increased expression of Bcl-2 and decreased that of Bax in H2O2-treated HUVECs. Moreover, caspase-3 activation induced by H2O2 was also decreased by propofol treatment in a dose-dependent manner. In agreement with the previously reported anti-apoptotic effects of propofol [82122], this study showed that propofol significantly protects HUVECs against apoptosis due to oxidative stress.
Many studies on vascular endothelial cells have demonstrated protective effects of propofol against oxidative stress; however, the intracellular signaling pathways have remained unclear. Among the signaling pathways in endothelial cells, MAPKs and Akt are essential for death and survival, respectively [23]. It has been reported that p38 MAPK acts as a pro-apoptotic signal while Akt serves as an anti-apoptotic signal in oxidativestress-induced endothelial cells [24]. These effects are mediated by apoptosis-related factors from the Bcl-2 family of proteins [25]. The effects of the JNK pathway in apoptosis remain controversial, since both pro- and anti-apoptotic effects have been reported, depending on the apoptotic stimuli and cell type in question [4]. Moreover, JNK can also phosphorylate both proand anti-apoptotic members of the Bcl-2 family [26]. Therefore, in the present study, we used three well-established components of apoptotic signaling pathways, p38 MAPK, JNK, and Akt, as indicators to verify the protective effect of propofol against H2O2-induced HUVEC apoptosis. Our findings suggested that pretreatment with propofol inhibited the phosphorylation of p38 MAPK, JNK, and Akt in H2O2-induced HUVECs.
It has been reported that propofol inhibits the phosphorylation of p38 MAPK in H2O2-treated HUVECs [2]. Similarly, we found that p38 MAPK was significantly activated by oxidative stress in cultured HUVECs, and propofol at 1-50 µM markedly inhibited p38 MAPK phosphorylation in H2O2-treated HUVECs. JNK phosphorylation is activated in various cell types, including endothelial cells [927]; however, the effect of propofol on JNK in H2O2-treated HUVECs has, to our knowledge, not been investigated. Similar to the p38 MAPK results, JNK phosphorylation was induced by H2O2 and suppressed by pretreatment with 25-50 µM propofol.
Akt is a critical regulator of cell survival that antagonizes mitochondria-directed apoptosis. Acute activation of Akt inhibits cell apoptosis induced by numerous stimuli, including H2O2 [8]. To determine whether Akt was the anti-apoptotic signaling pathway that mediates the protective role of propofol in HUVECs, we evaluated H2O2-induced phosphorylation and activation of Akt at Ser473. Treatment with H2O2 for 4 h markedly increased Akt phosphorylation, and pretreatment with 25-50 µM propofol for 30 min dramatically inhibited the H2O2-induced Akt phosphorylation in HUVECs. This finding was unexpected and seems to contradict the results of other studies [3828]. Cell types, extracellular conditions, the altered cellular redox status and/or timing of Akt measurement may explain this discrepancy. Treatment of different cell types with various concentrations of H2O2 for different periods yields inconsistent results [38]. Additionally, the Akt pathway was fully activated at ~10 min after H2O2 treatment, and then declined gradually [8]. Regretfully, we did not measure Akt expression at 10 min, but assayed it at 4 h after H2O2 treatment.
Several limitations to this study should be noted. Firstly, this study used only pretreatment with propofol for 30 min before co-incubation with H2O2. Post-treatment with low concentrations of propofol (e.g., 1-10 µM) is closer to the actual levels used for ICU long-term sedation in critically ill patients. Secondly, further studies using specific signal pathway inhibitors and measurement of the levels of proteins involved in many signaling pathways for various periods are required to elucidate the effects of propofol on endothelial cell apoptosis.
Propofol protected HUVCEs from H2O2-induced apoptosis in a dose-dependent manner. The mechanism may involve the p38 MAPK, JNK, and Akt signaling pathways. These findings suggest that propofol is a preemptive anesthetic in patients with cardiovascular disease such as hypertension, atherosclerosis, and diabetic vasculopathy.




 XML Download
XML Download