

 PDF
PDF Citation
Citation Print
Print


Introduction and Background
Subarachnoid hemorrhage (SAH) cases represent approximately 7% of all types of stroke], and have a notoriously high mortality rate [1]. Despite fluctuations, the overall incidence of SAH has been stable at around 10 per 100,000 population per year in most countries for the past few decades [2,3]. However, in one region of Japan, the incidence of SAH was reported to be as high as 23-25 per 100,000 people per year, one of the highest worldwide [2]. It is possible that this extraordinary incidence might be the consequence of more assiduous diagnosis by early computed tomography (CT). Mortality after SAH is directly related to the severity of illness, and functional outcomes after SAH vary widely. The age, sex, and grade-adjusted overall 30-d fatality rate for SAH is about 45-50% [4,5,6,7].
Preoperative, Initial Phase: ABC and EVD
Critical care requirements could differ depending on a patient's presentation based on the initial Hunt and Hess scale, which can be graded by performing a careful clinical assessment of the patient's neurologic status as follows:
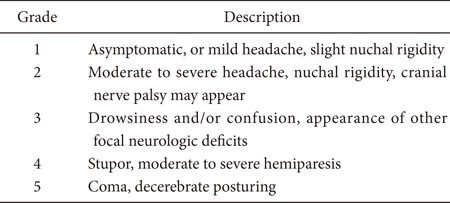
When SAH is at a low grade (Hunt and Hess SAH grade 1-2), perioperative monitoring and management in the intensive care unit (ICU) is quite straightforward. It consists of hemodynamic and cerebral assessment at the bedside, routine cardiac monitoring, careful and repetitive clinical examination, and basic blood work. The risk of symptomatic vasospasm is low, and when it does develop, the neurological change is clinically obvious. Nonetheless, a low grade of neurological injury may become acutely worse when it is complicated by acute vasospasm that induces symptomatic acute and/or delayed cerebral ischemia. A characteristic example is a patient who initially presents with an alert, oriented neurologic status (low grade Hunt and Hess status). However, the patient has a diffuse, thick subarachnoid blood collection consistent with classic Fisher scale 3, which may induce sudden and rapid deterioration. Most of the time, these patients do not require tracheal intubation but must be closely followed with frequent neurological examination by the ICU nursing and physician staff.
A high grade of SAH is evidenced by Hunt and Hess grade 3-5, a large amount of subarachnoid and intraventricular hemorrhage, radiographic evidence of hydrocephalus, and deteriorating mental status. This is a considerably more challenging situation, and the fundamental goal is to maintain adequate cerebral perfusion pressure (CPP). The CPP is the difference between the mean arterial blood pressure (MAP) and the intracranial pressure (ICP):
Initially, these patients develop very labile intracranial hypertension, a harbinger of potentially impaired CPP. As always, the first step is to ensure adequate resuscitation and control of airway, breathing, and circulation (ABC). Next, an external ventricular drain (EVD) should be placed to ensure continuous ICP monitoring. Then, to maintain adequate CPP and avoid further injury to the brain tissue, the MAP must be adjusted relative to the ICP.
Unfortunately, there is little evidence to promote an ideal CPP, which is not only unpredictable, but which, because the disease is so dynamic, may fluctuate widely from day to day. In general, it is preferable to keep the MAP under tight control while maintaining a CPP of 50 to 70 mmHg. This mandates keeping the ICP less than 20 mmHg, with MAP 70-90 mmHg and systolic blood pressure (SBP) preferably constrained to less than 140 mmHg. Tight blood pressure control is essential until the ruptured aneurysm is fully secured.
Proactive Management for the Postoperative Period - A Paradigm Shift in Goal-directed Therapy
A recently introduced paradigm shift toward aggressive protocolized management has demonstrated that mortality in patients with Hunt and Hess grade 4 and 5 SAH can be significantly reduced compared with historical controls [9].
The paradigm is initiated by emergent rescue of SAH victims with ABC support, followed by immediate admission to a highly specialized neurointensive care unit. The fundamental principles of management include early aggressive medical and surgical therapy and the use of advanced technologies to monitor the patient's injured brain. Recommended interventions include immediate placement of an external ventricular drain, timely securing of the ruptured aneurysm, prophylaxis against rebleeding, control of elevated ICP, and optimization of CPP. In a sense, the key is to provide a physiologically optimal environment to promote recovery of the injured neurons. The paradigm also encompasses compassionate end-of-life care without premature withdrawal of life support [9].
The most meaningful way to follow a brain-injured patient is to perform serial bedside neurological examinations. Unfortunately, with high-grade SAH, there is no examination to follow - the patient may remain completely comatose for days or weeks, and the only clinical finding of any portent may be that of intact brainstem reflexes. This frequently leads the clinicians to disclose a dismal prognosis and likelihood of extremely poor outcome to the family, which in turn understandably culminates in withdrawal of medical support.
Becker et al. [10] suggest that clinicians who use initial data to predict outcome may be overly pessimistic, leading to possibly premature withdrawal of support and a "self-fulfilling prophecy". Traditionally, clinicians tend to react to their observations of patients in the ICU. For example, if the MAP decreases, an assessment of intravascular volume status is initiated, quickly followed by a combination of fluid administration and vasopressor therapy with or without inotropic support. If arterial oxygen saturation (SaO2) falls, an investigation of possible etiologies begins and a treatment algorithm follows based upon the findings. Such "reactive" treatment strategies may be successful for some disease processes, but they do not achieve the best results in advanced-grade SAH. The patient's brain is severely injured, and there is an intense, active inflammatory reaction to the presence of blood in the subarachnoid space [11,12]. The clinical examination of a stuporous or comatose patient typically remains unchanged for a protracted length of time. When meaningful changes finally occur, it may be too late to mount an effective therapeutic response.
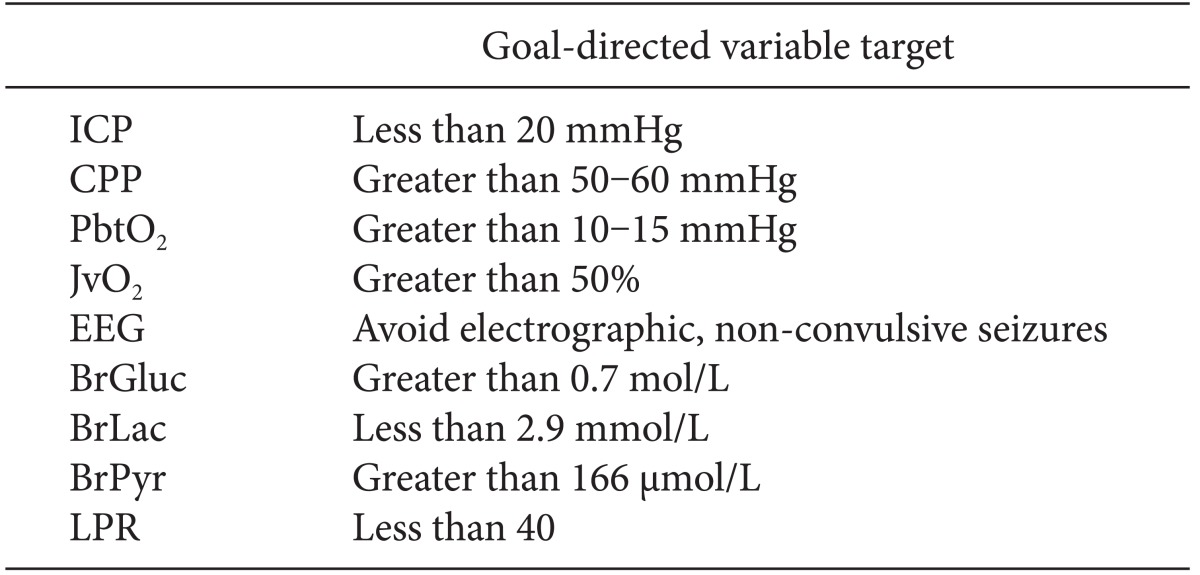
The paucity of meaningful information from clinical examination in patients with high-grade SAH has driven the development of multi-modal monitoring and goal-directed therapy (Table 1). In this approach, the daily neurological examination is augmented by continuous electroencephalography (EEG), real-time measurement of partial pressure of brain oxygen tension (PbtO2), and cerebral microdialysis. The clinician utilizes input from multiple variables proactively to adjust patient hemodynamics and achieve the desired level of brain perfusion, which may vary day-to-day or even hour-to-hour. In this way, the entire ICU team works toward a clearly defined, common goal. Although there is not sufficient data to support the idea that any of these goal-directed therapies might lead to improved outcome, these targets are physiologically reasonable goals until further data is available.
For example, the following monitoring goals could be set at any given time: keep ICP < 20 mmHg, CPP > 60 mmHg, and PbtO2 > 10 mmHg. The primary mechanism to achieve these goals is continuous assessment and correction of cardiac output and performance, hemoglobin concentration, and intravascular volume status. Successful achievement of these goals implies adequate pulmonary gas exchange, avoidance and prompt treatment of fluid overload (and risk of pneumonia), and optimal cerebral oxygen delivery. This in turn provides a physiologically optimal environment to the injured brain, giving it the best chance for ultimate recovery.
Intensivists devote a considerable portion of each day to attempting to determine the intravascular volume status of critically ill patients through the use of clinical examination and standard parameters that are often quite misleading [13]. The mainstay of assessment, CVP, is a stagnant pressure measurement that is criticized for its unreliable value as a surrogate for circulating blood volume or preload responsiveness in the operating room or ICU [14]. It therefore seems logical to use more than one hemodynamic value to determine the appropriate fluid therapy. This rationale has provided the impetus for the introduction of semi-invasive, pulse contour methods of continuous hemodynamic monitoring, highlighted by Doppler ultrasonography [15,16]. For example, insonation of the inferior vena cava diameter and its collapsibility correlates quite well with other parameters and provides dynamic assessment of intravascular fluid status in critically ill patients[17,18,19].
Regardless of which variables one follows, it is of paramount importance to ensure a patient condition that is as close to euvolemia as possible. Severe hypovolemia must be avoided at all costs. The severe decrease in cerebral blood flow that follows is detrimental to the injured brain, especially during the vasospastic period, and is associated with an unacceptably high risk of cerebral infarction. At the other extreme, volume overload (which includes prophylactic hypervolemic therapy even before a patient suffers any symptomatic vasospasm) may increase urine output and cardiac filing pressures, but may not provide any benefits in cerebral perfusion [20]. When hypervolemia results in pulmonary congestion and edema, arterial hypoxemia further compromises oxygen delivery to the injured brain.
Critical Care for Symptomatic Cerebral Vasospasm
The ICU team should provide 24-7 coverage for SAH patients, preferably in a dedicated neurointensive care unit, because these patients require continuous, round-the-clock neurological monitoring. At the first sign of symptomatic cerebral vasospasm, specific treatment must be initiated without delay. Current standards of care mandate that this should occur within 2 h [21].
While there is considerable variability among clinicians in therapy for cerebral vasospasm, certain principles are accepted by all.
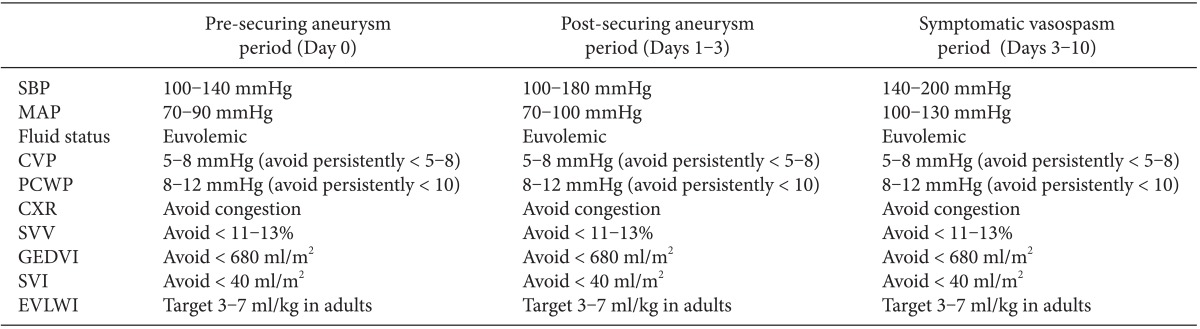
First, it is mandatory to avoid volume depletion and systemic hypotension, which decrease cerebral perfusion and increase the risk of cerebral ischemia. At the onset of symptomatic vasospasm, it is essential to assure euvolemia and preferable to initiate hypertensive therapy. Elevation in systemic blood pressure increases perfusion of the vasospastic, ischemic brain. While so-called "triple-H" therapy (hypervolemia, hypertension, and hemodilution) has never been shown to improve long-term outcome in any randomized controlled settings [22], it is reasonable to initiate interventions that assure euvolemia and mild to moderate systemic hypertension. A representative example of hemodynamic targets is outlined in Table 2.
There is no evidence to support prophylactic hypervolemic therapy or induction of hypertensive therapy prior to the onset of symptomatic vasospasm. A recent survey performed by the Neurocritical Care Society (NCS) revealed that medical centers without a dedicated neurointensive care unit are more likely to use prophylactic hypervolemia [23]. All 375 members of the NCS reported that they agree with the induction of hypertension for severe or symptomatic vasospasm.
The options for endovascular and neurosurgical intervention in patients with symptomatic cerebral vasospasm are beyond the scope of this article and will not be discussed further here.
Paroxysmal Sympathetic Hyperactivity: A Devastating Complication
Acute SAH is not uncommonly accompanied by an excess of sympathoadrenal activity that is described by a variety of terms, including paroxysmal sympathetic hyperactivity (PSH), central nervous system storming, autonomic storming, and dysautonomic hyperactivity, among others. Its exact pathophysiology has not been elucidated, and this syndrome is not confined to SAH. It has been observed after a variety of other sources of acute brain injury including traumatic brain injury, intracranial hemorrhage, anoxic brain injury, and encephalitis. Rarely, acute parasympathetic reactions have been reported.
PSH has a sudden onset without warning signs or obvious triggers. It is characterized by hypertension, sinus tachycardia, hyperventilation, facial flushing, hyperemia, diaphoresis, hypertonia, hyperreflexia, and fever [24]. When patients with SAH develop PSH, their course is substantially worsened. They have prolonged ICU and hospital stays, increased medical complications, and poor long-term functional outcomes.
A rational critical care approach includes prompt diagnosis, the ruling out of other underlying medical etiologies, and the initiation of appropriate medical therapy. A number of different agents have reported to be useful in PSH patients, including benzodiazepines, bromocriptine, opioids, beta-blockers, baclofen, alpha-2 agonists, and antiepileptic drugs. It is intuitive that these patients would benefit from deep sedation, intubation, and mechanical ventilation with high-dose IV midazolam or propofol. However, outcomes appear to be enhanced by minimizing sedation so that patients can stay awake, remain extubated, and ultimately move out of the ICU. Maintaining the balance between adequate pharmacological treatment and avoidance of excessive sedation is challenging but crucial [24,25].
Neurogenic Cardiomyopathy and Transient Apical Ballooning Syndrome
A variable percentage of patients with high-grade SAH may present with acute heart failure, with dyspnea, substernal chest pain, pulmonary congestion and edema, and systolic hypotension. Electrocardiographic changes are quite common even without myocardial dysfunction, and include ST elevation, inverted T waves, and prolonged QT interval. A wide variety of arrhythmias may occur, especially within the first 48 h. These include sinus bradycardia, supraventricular tachycardia, atrial fibrillation, ventricular ectopy and tachycardia, and in the presence of prolonged QT interval, polymorphic ventricular tachycardia (torsades de pointes).
Echocardiography reveals impaired myocardial contractility and a substantially decreased ejection fraction, often to less than 25%. Cardiac enzymes such as troponin are elevated. This condition may be characterized as an acute neurogenic cardiomyopathy, akin to the stunned myocardium encountered after ischemia-reperfusion injury.
A considerable proportion of patients, mostly postmenopausal women, develop a specific variant that causes ballooning of the ventricular apex with sparing of the basal segments [26]. Because of the characteristic shape of the ventricle, this syndrome is often called takotsubo cardiomyopathy, after the Japanese for an octopus trap, or more formally, transient left ventricular apical ballooning syndrome. It is associated with prolonged tracheal intubation and mechanical ventilation and cerebral vasospasm. This phenomenon, however, has been described in all age groups and both sexes.
Care for these patients is largely supportive, and the primary goal is to minimize myocardial oxygen consumption and maximize myocardial perfusion. This includes prompt treatment of cardiac arrhythmias, sedation and mechanical ventilation, aspirin, nitrites, statins, beta-blockers, calcium channel blockers, and angiotensin-converting enzyme inhibitors. Potent inotropic agents should be avoided if possible, because they increase myocardial oxygen demand. Judicious use of a non-catecholamine alpha-adrenergic agent, phenylephrine, is preferred to treat severe systemic hypotension, but in severe cases, intra-aortic balloon counterpulsation may be required to assure adequate myocardial perfusion pressure and decreased myocardial demand [27]. Clearly, the use of pure vasopressors without any inotropic properties should also be avoided as this increases the afterload and could potentially make heart failure worse. As with any hypotensive case, the first step would be to assess the volume status rather than jumping to start vasopressors to elevate the blood pressure. Typically, the syndrome is reversible after 3-5 d as long as no further injury occurs to the myocardium.
Summary and Conclusion
The perioperative management of patients with SAH is challenging, especially when the grade is high. Typically, the initial medical battle is to deal with critical intracranial hypertension and cerebral edema ("ICP crisis"). This is followed by what may be life-threatening cerebral vasospasm identified by angiography or ultrasonography, preferably before it becomes symptomatic. After a struggle with vasospasm, decisions loom regarding placement of a ventriculo-peritoneal shunt versus weaning the EVD. If the patient survives the first 1 or 2 weeks, PSH may occur at any time and without any warning. Improving the long-term outcome after SAH truly requires a well-trained, experienced ICU team, preferably in a specialized neurointensive care unit, to provide optimal monitoring and timely and appropriate therapy. A multidisciplinary effort is needed to promptly diagnose and treat the multiple challenges of the dynamic disease that is SAH.

 XML Download
XML Download