

 PDF
PDF Citation
Citation Print
Print


Introduction
Nitric oxide (NO) constitutively produced by endothelial nitric oxide synthase (eNOS) plays a major role in the regulation of blood pressure, vascular tone, and serves to protect against cardiovascular disease [1]. A number of studies have demonstrated that NO plays a modulating role in the post-infarction remodeling process, which involves a modest but lasting upregulation of eNOS in blood vessels and cardiomyocytes [2-5]. In contrast, inducible NOS (iNOS) showed dramatic de novo formation 1 week after infarction, predominantly in the infarcted area and cardiomyocytes [5-7]. Moreover, a gradually increased myocardial production of superoxide (O2-) has been detected during remodeling in the peri-infarcted and remote myocardium [5,8,9]. The reaction of superoxide with NO reduces the bioavailability of NO as a vasodilator by generating peroxynitrite (a product of NO + O2-), which itself may contribute adversely to vascular function and the compensatory effects of NO and thereby influence post-infarction remodeling [8,9]. Therefore, vascular reactivity at the early stage after acute myocardial infarction (AMI) may be changed by several mechanisms, such as enhanced eNOS or iNOS activity, or the reduction of bioactive NO by superoxide.
Some studies have demonstrated that the change of vascular reactivity during the post-infarction remodeling process can occur at non-cardiac vessels such as the large conduit artery or resistant artery [7,10]. However, the effects of vascular contractile responses during the post-infarction remodeling process are determined by the underlying mechanisms. Some reports indicate that the activity of iNOS produces increased α1-adrenergic receptor (AR)-mediated contraction by phenylephrine (PE) in rat caudal vascular beds 3 days after AMI [7]. Other studies suggest that enhanced eNOS activity can play an important role in mediating the reduced vascular growth and decreased PE-induced contractions [10,11].
PE-induced contraction involves various calcium entry mechanisms or channels such as L-type voltage-operated calcium channels (VOCCs), receptor-operated calcium channels (ROCCs), capacitative calcium entry (CCE) by the activation of store-operated calcium channels (SOCCs), reversal mode of sodium-calcium exchangers (NCX), and non-capacitative calcium entry (NCCE) via the activation of diacyl glycerol (DAG) lipase [12-17]. Recent findings indicate that some calcium entry mechanisms can be affected by endothelial NO, which can inhibit VOCCs or SOCCs [18]. However, it has not been determined which calcium channels are changed in rat aorta 3 days after AMI. Therefore, we tested the hypothesis that the role of each calcium channel or relative contribution of calcium entry mechanisms may change or differs in rats 3 days after AMI. Based on several previous reports concerning rat aorta [10,11], we investigated calcium entry mechanisms of vascular smooth muscle after AMI and tested the effect on PE-induced contraction using the SOCC inhibitor 2-aminoethoxydiphenyl borate (2-APB), a SOCC inducer using thapsigargin (TG), the NCCE inhibitor RHC80267, and the selective NCX inhibitor 3,4-dichlorobenzamil hydrochloride (3,4-DCB). Finally, we obtained dose-response curves to the VOCC inhibitor nifedipine to determine the relative contribution of each calcium channel or calcium entry mechanism to PE-induced contraction.
Materials and Methods
All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee of the Medical Center.
Preparation of the AMI model
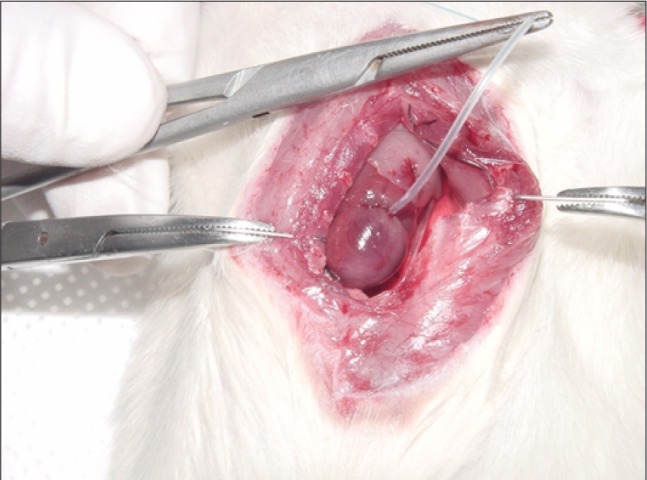
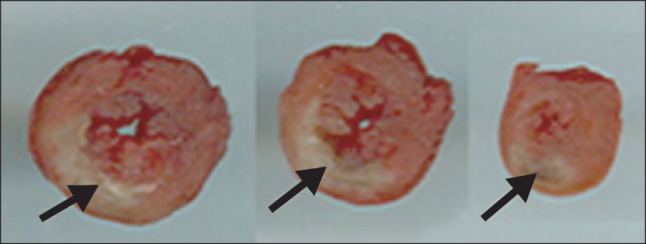
Male Sprague Dawley rats (8 to 9 weeks old) weighing 280 to 330 g were anesthetized with administration of ketamine (80 mg/kg) intramuscularly. Rats were placed in either the AMI or sham-operated (SHAM) group. In brief, rats were anesthetized with ketamine and subjected to median sternotomy. The heart was exteriorized and the left anterior descending coronary artery (LAD) was then surrounded with 6-0 nylon in the AMI group. The loop around the LAD was tightened for 30 minutes and then released to induce AMI (Fig. 1). In the sham groups, the same operation was performed without LAD occlusion. The heart was then returned to its original position and the incision was closed. The left ventricle was cut into three or four slices transversely from base to apex 3 days after AMI or the sham operation. The slices were incubated with 2,3,5-triphenyl-tetrazolium-chloride (TTC) for 10 minutes. Non-infarcted myocardium, which contained dehydrogenase, was stained brick red by reacting with TTC, whereas necrotic (infarcted) tissue was unstained because of the lack of enzyme [10].
Preparation of aortic rings for tension measurement
The descending thoracic aorta was dissected free and cut into aortic rings each with a length of 4-5 mm 3 days after AMI or the sham operation. All rings were immersed in cold modified Krebs-Ringer bicarbonate (KRB) solution with the following composition (mM): 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.4 CaCl2, 25 NaHCO3, 11.1 glucose, and 0.016 EDTA. After removing connective tissue, the aorta was cut into ring segments 5 mm in length, with care taken not to damage the endothelium. In some rings, the endothelium was intentionally denuded by gently rubbing the inner surface with a cotton swab.
Isometric tension experiments
Aortic rings were vertically suspended between two steel hooks in an organ chamber filled with 10 ml of modified KRB solution gassed with 95% O2 and 5% CO2. The temperature of the organ bath was controlled with a refrigerated bath circulator (RBC-10, Jeio Tech, Seoul, Korea). One of the hooks was anchored and the other was connected to a strain gauge (FT-03, Grass Instruments, Quincy, MA, USA) to measure the isometric tension. Rings were stretched at 10 min intervals in increments of 0.5 g to reach the optimal tension. The optimal tension was defined as the minimum level of stretch required to achieve the largest contractile response to 60 mM KCl, and was determined in a preliminary experiment to be 2.0 g for the size of aortic rings used in these experiments. After the rings had been stretched to their optimal resting tension, the contractile response to 60 mM KCl was measured which shows the values of no drug rings in the results. After washing out the KCl from the organ bath and returning the isometric tension to pre-stimulation values, each ring was pre-contracted with the α1-AR agonist PE (10-7 M) and the relaxation response to acetylcholine (10-6 M) was recorded to assess endothelial integrity. Endothelium-intact rings were verified by a relaxation greater than 50% in response to acetylcholine, whereas denudation was recognized by a relaxation of less than 5%.
The first series of these in vitro experiment with KRB containing 2.5 mM Ca2+ was conducted to assess the contractile responses induced by PE in endothelium-intact or denuded rings in SHAM and AMI groups. After determining endothelial integrity, cumulative concentration-response studies for PE (10-9 to 10-5 M) were performed in both groups.
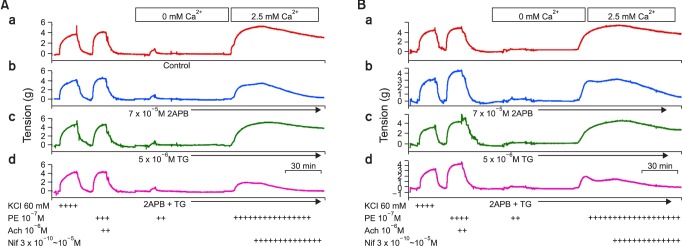
The second series of experiments were designed to determine which calcium channels or calcium entry mechanisms were responsible for the PE-induced contraction in the AMI group. Endothelium-denuded rat aortic rings were treated with calcium-free buffer containing 2 mM ethylene glycol tetraacetic acid (EGTA) for 10 min and then replaced with calcium-free buffer without EGTA. After 10 min, this solution was replaced with calcium-free buffer containing PE (10-7 M). When the KRB solution containing 2.5 mM Ca2+ was replaced, ongoing tonic contraction induced by PE was assessed in both groups. To clarify the role of SOCCs on PE-induced contraction, we investigated PE-induced contraction in rings pretreated with inositol 1,4,5-trisphosphate receptor (IP3R) blocker or SOCC blocker 2-APB (7.5 × 10-5 M), and sarco/endoplasmic-reticulum Ca2+ ATPase (SERCA) inhibitor or the SOCC inducer TG (5 × 10-6 M). In addition, we used RHC80267, a selective inhibitor of DAG lipase, to prevent the activation of NCCE by PE. We also used the selective NCX inhibitor 3,4-DCB (10-4 M) to elucidate the role of NCX on PE-induced contraction in both groups. Finally, we obtained dose-response curves to the VOCC inhibitor nifedipine (3 × 10-10~10-5M). When ongoing tonic contraction by PE (10-7 M) was sustained, cumulative dose-response relationships of nifedipine were obtained and compared between the two groups, or under conditions of SOCC inhibition with 2-APB or SOCC induction with TG.
Drugs and solutions
All drugs were commercially available and of the highest purity: PE, acetylcholine, nifedipine, TG, 2-APB, RHC80267, 3,4-DCB, and EGTA (Sigma Chemical, St. Louis, MO, USA). The final concentration of dimethyl sulfoxide in the study chamber was less than 0.1% (vol/vol). All other drugs were dissolved and diluted in distilled water. All drug concentrations were expressed as the final molar concentration in the organ bath.
Data analysis
All data are expressed as mean ± SEM. Contractile responses to PE and calcium are expressed as grams (g) of absolute tension. The maximum contraction or relaxation (Rmax) was considered to be the maximal amplitude of the response reached in concentration-response curves to contractile or vasorelaxing agents, respectively. The logarithm of the drug concentration eliciting 50% of the maximal contractile or vasorelaxing response (pEC50%) was calculated using non-linear regression analysis by fitting the concentration-response relation for PE to a sigmoidal curve using commercially available software (Prism version 4.0; Graph Pad Software, San Diego, CA, USA). Statistical analysis for comparison of the pEC50% and Rmax values of each drug was performed with the one-way analysis of variance (ANOVA) test followed by Fisher's least significant difference method using SPSS software (ver. 17.0 for Windows; SPSS, Chicago, IL). Differences were considered statistically significant for P values < 0.05. N refers to the number of rats whose descending thoracic aortic rings were used in each protocol.
Results
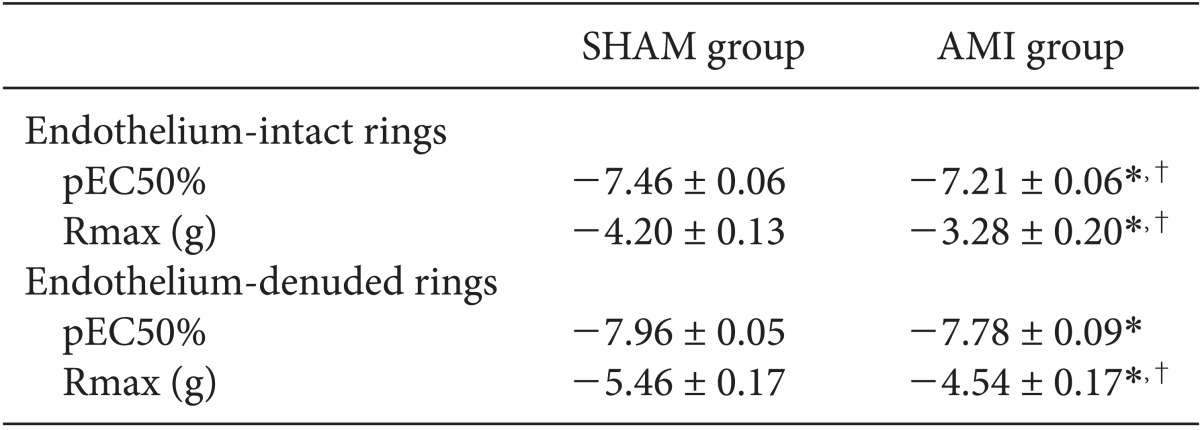
Dose-response relationships of PE
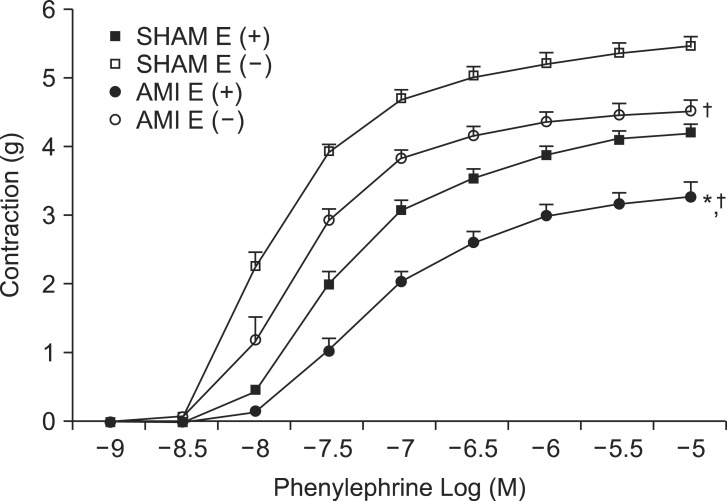
PE dose-response relationships of endothelium-intact rings in the AMI group shifted to the right (Table 2, Fig. 3). pEC50% and Rmax of PE for endothelium-intact rings of the AMI group differed significantly (P < 0.05) from that of endothelium-intact rings of the SHAM group. Rmax of endothelium-denuded rings in the AMI group was significantly lower (P < 0.05) than that of endothelium-denuded rings in the SHAM group.
Effects of SOCC activation or inhibition on PE-induced contraction
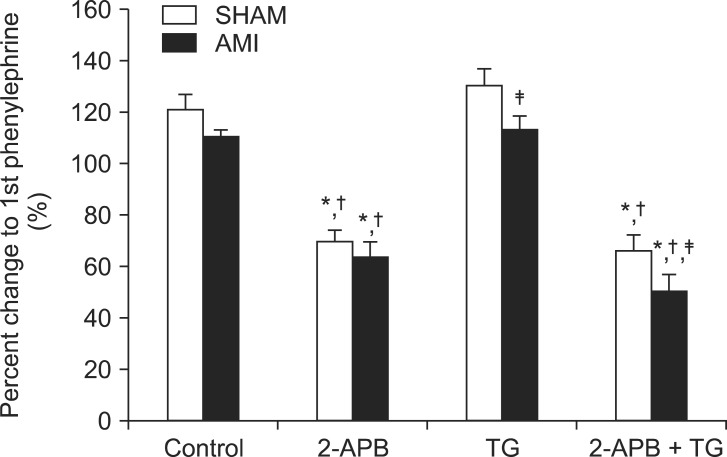
PE-induced contraction in a 2.5 mM Ca2+ medium in the AMI group was slightly, but not significantly (P > 0.05), attenuated in endothelium-denuded aortic rings of the AMI group (Fig. 4, n = 6). SOCC inhibition with 2-APB (7.5 × 10-5 M) significantly attenuated (P < 0.05) PE-induced contraction in both groups. SOCC induction with TG (5 × 10-6 M) had no marked effect on PE-induced contraction. However, there were statistical differences (P < 0.05) in PE-induced contraction in TG-pretreated rings with or without 2-APB between the two groups.
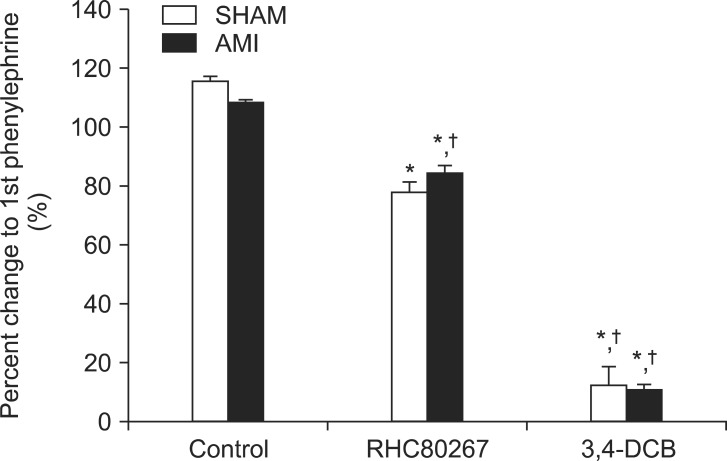
Effects of DAG lipase inhibition on PE-induced contraction
To assess the relative contribution of NCCE, we investigated the effects of a selective DAG lipase inhibitor on PE-induced contraction. DAG lipase inhibition with RHC 80267 (5 × 10-5 M) significantly attenuated (P < 0.05) PE-induced contraction (Fig. 5, n = 4). However, there were no differences (P > 0.05) between the two groups.
Effects of NCX inhibition on PE-induced contraction
The selective NCX inhibitor 3,4-DCB (10-4 M) was used to investigate the role of NCX on PE-induced contraction. Our findings showed that 3,4-DCB completely abolished PE-induced contraction in both groups (Fig. 5, n = 4). However, there were no differences (P > 0.05) between the two groups.
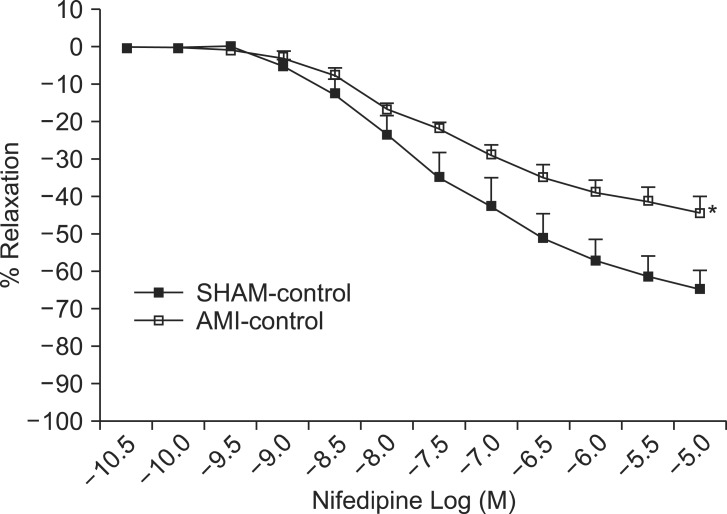
Effects of L-type VOCC inhibition on PE-induced contraction
To evaluate the relative contribution of VOCCs, we measured the dose-response relationships of nifedipine when PE-induced contraction was sustained. The dose-response relationships of nifedipine in the AMI group shifted to the right (Fig. 6). Rmax of nifedipine in the AMI group was significantly lower (P < 0.05) than that of the SHAM group but pEC50% was not significantly different.
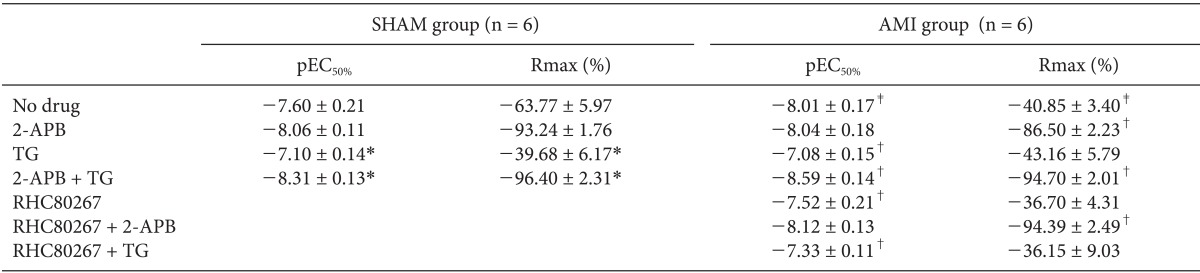
Effects of L-type VOCC inhibition under various conditions
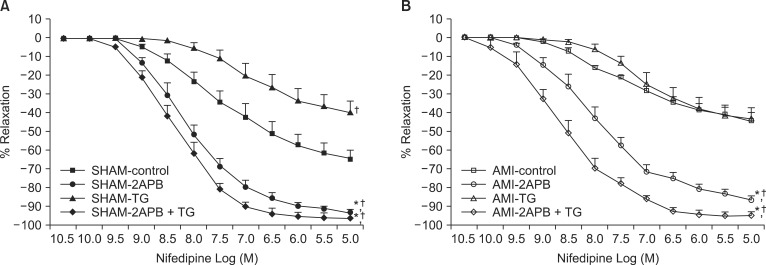
Fig. 7 shows the original tracing of the dose-response relationships of nifedipine (3 × 10-10~10-5 M) in SHAM (A) and AMI (B) groups after restoration of 2.5 mM Ca2+ and PE (10-7 M), which were measured under various conditions (Fig. 8, Table 3). The cumulative addition of the VOCC blocker nifedipine produced a dose-dependent vasorelaxation in endothelium-denuded control rings (Fig. 8A, n = 6). These vasorelaxing effects of nifedipine were significantly potentiated under conditions of SOCC inhibition with 2-APB (7.5 × 10-5 M) in both groups. However, these effects were significantly attenuated under conditions of SOCC induction with TG in the SHAM group. In contrast, the attenuating effects induced by TG did not appear in the AMI group (Fig. 8B, n = 6). Moreover, 2-APB significantly potentiated nifedipine-induced vasorelaxation in rings treated with TG in the AMI group. Nifedipine-induced vasorelaxation of rings in the AMI group treated with the DAG lipase inhibitor RHC80267 did not differ from that of control rings (Table 3).
Discussion
We demonstrated in this in vitro study the decreased sensitivity (pEC50%) and efficiency (Rmax) of PE in endothelium-intact rings in 2.5 mM Ca2+ medium 3 days after AMI. We also found that the effect of SOCC induction with TG pretreatment in 0 mM Ca2+ medium on PE (10-7 M)-mediated contraction after the restoration of 2.5 mM Ca2+ was significantly lower in endothelium-denuded rings of the AMI group than the SHAM group. Moreover, we demonstrated decreased pEC50% and Rmax for the VOCC inhibitor nifedipine on PE-mediated contraction, suggesting that VOCC-independent calcium entry mechanisms play a major role in PE-mediated contraction in rat aorta of the AMI group. Finally, we demonstrated the enhanced CCE pathway via the activation of SOCCs involved in these enhanced VOCC-independent calcium entry mechanisms in the AMI group.
As in previous in vitro studies with rat aorta [10], our results support the assertion that vascular contractile responses in a large conduit artery can be decreased at the early stage after myocardial ischemic reperfusion injury or AMI. In the current study, pEC50% and Rmax of PE in endothelium-intact rings of the AMI group decreased compared with those of the SHAM group, whereas only Rmax of PE in endothelium-denuded rings decreased significantly in the AMI group. These results suggest that endothelium-dependent mechanisms may be involved in the decreased sensitivity and efficiency for PE in rat aorta 3 days after AMI. Previous research demonstrated that these findings were associated with the up-regulation of NO-cyclic guanosine monophosphate (cGMP) pathways, which was supported by enhanced eNOS expression, increased NO metabolites and the basal cGMP concentration [10]. In addition, the NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME) inhibited these decreased PE-induced contractions in the AMI group. The overall findings clearly indicate that the vascular contractile response during an early stage of the post-infarction remodeling process can be affected by the enhanced eNOS activity [10,11].
To investigate other possible mechanisms responsible for the change of vascular reactivity in rat aorta in the post-infarction remodeling process, we focused on calcium entry mechanisms that are associated with three calcium channels (SOCCs, VOCCs, reversal mode of NCX). These calcium channels are well known to be involved in PE-induced contraction [14]. PE stimulates phospholipase C (PLC) leading to formation of InsP3 and DAG, each of which leads to activation of a distinct calcium entry pathway [14,19]. InsP3 activates InsP3R and stimulates the release of calcium from intracellular stores and thereby generates the signal required for activation of SOCCs, which is known as the CCE pathway [19,20]. This CCE pathway can also be activated by emptying the intracellular stores using TG and is selectively blocked by 2-APB (100 µM) [21,22]. In addition, arachidonic acid, produced from DAG lipase, activates another calcium entry pathway [16,17]. This NCCE pathway is permeable to calcium and is blocked by RHC 80267, a selective inhibitor of DAG lipase [17]. PE also produces calcium influx by depolarization, which is evoked by the opening of VOCCs and the reverse mode of NCX [15,23]. Since the absence of selective blockers for ROCCs and CCE has strongly hampered their distinction from other calcium transporting mechanisms and thus prevented a clear understanding of their roles in regulating smooth muscle functions, we tested the involvement of one calcium entry mechanism when other calcium entry mechanisms were blocked with their selective blockers.
SOCCs are involved in the CCE pathway and are important for sustaining the tension mediated by PE [20]. We also found that the effect of SOCC induction with TG pretreatment in 0 mM Ca2+ medium on PE (10-7 M)-induced contraction after the restoration of 2.5 mM Ca2+ was significantly lower in endothelium-denuded rings of the AMI group compared to the SHAM group. Since this effect of TG can be blocked by 2-APB, which is known as a SOCC blocker, it is possible that SOCCs in the AMI group are already activated and therefore SOCC induction with TG has no effect, or no further effect, on PE-induced contraction. Furthermore, although these findings also suggest the occurrence of an enhanced CCE pathway on PE-induced contraction in the AMI group, we could not confirm the occurrence of an enhanced CCE pathway on PE-induced contraction on the basis of the TG results. To distinguish the CCE pathway from other calcium transporting mechanisms, calcium entry via VOCC-dependent calcium entry mechanisms or other possible calcium entry pathways must be specifically inhibited by their selective blockers. L-type VOCCs provide a portion of the calcium used to refill the sarcoplasmic reticulum (SR) calcium store and to sustain tonic contraction. Based on these considerations, we obtained nifedipine dose-response relationships to investigate the involvement of VOCC-independent calcium entry mechanisms on PE-induced contraction. Our results demonstrated that the VOCC inhibitor nifedipine produced a dose-dependent inhibitory effect on PE-induced contraction in both groups, but pEC50% and Rmax of rings with nifedipine were significantly lower in the AMI group compared to the SHAM group. These findings suggest the decreased role or contribution of VOCCs to PE-induced contraction in the AMI group. We think these findings are associated with enhanced NO activity during the post-infarction remodeling process [4,5,9]. Recent investigation has shown that NO was involved in the blocking of L-type calcium influx via the NO-cGMP pathway in mouse aorta [18]. Furthermore, a previous study indicated that the hypo-responsiveness for PE in the AMI group was associated with the up-regulation of eNOS expression and activity [10].
In the current study, we demonstrated that the enhanced CCE pathway via the activation of SOCCs plays a central role on these VOCC-independent calcium entry mechanisms in the AMI group. This is also supported by other evidence obtained in the current study. First, pEC50% and Rmax of nifedipine in control rings of the AMI group were significantly lower than those of the SHAM group, suggesting that VOCC-independent calcium entry mechanisms play a more important role on PE-mediated contraction in the AMI group than in the SHAM group. Second, there were no differences in Rmax for nifedipine between control rings and TG pretreated rings in the AMI group, whereas there were significant differences in Rmax for nifedipine between control rings and TG pretreated rings in the SHAM group, indicating that VOCC-independent calcium entry mechanisms in the AMI group are similar to those of rings with SOCC induction by TG. Third, pEC50% and Rmax of nifedipine under conditions of SOCC inhibition by 2-APB were significantly higher in the AMI group, suggesting that VOCC-independent calcium entry mechanisms can be blocked by 2-APB. In addition, 2-APB also produced the same effect under conditions of SOCC induction by TG. These overall findings indicate that the VOCC-independent calcium entry mechanisms in the current study were induced by an enhanced CCE pathway through activation of SOCCs.
Since calcium efflux from SR in vascular smooth muscle is mediated by two important calcium permeant channels comprising InsP3Rs and ryanodine receptors, we considered the effects of the InsP3R blocker 2-APB on PE-induced contractions. The InsP3Rs on the SR play a crucial role by releasing calcium to activate the myosin light chain kinase units tethered to the myofilaments [24]. The finding that PE-induced contraction was significantly attenuated by 2-APB, which is basically known as an InsP3R blocker, suggests that the opening of InsP3R channels is required for PE-induced contractions in the rat aorta. In addition, the findings in the current study showing that PE-mediated contractions in rings pretreated with TG were significantly attenuated by 2-APB suggest that activation of CCE mediated by TG can be blocked by 2-APB. In fact, 2-APB can block the CCE pathway in several other cells when SR depletion is maintained by a SERCA blockade. Although 2-APB has been known as a specific membrane-permeable InsP3R antagonist, emerging data show that its primary action on cells is not to block calcium release, but rather to inhibit CCE. The importance of 2-APB as demonstrated by the involvement of InsP3R coupling to SOCCs [25] is highlighted by the finding that 2-APB can inhibit SOCCs directly without involvement of InsP3Rs [26,27]. Despite its widespread use, there is presently no clear-cut evidence for 2-APB inhibiting calcium signaling by solely targeting InsP3Rs. Therefore, at best, it is a reasonable interpretation that 2-APB can inhibit both agonist-induced calcium release and the concomitant SOCCs with the same efficacy as found in the current study.
The activation of non-selective cation channels (NSCC; e.g., ROCCs/SOCCs) can produce mainly an influx of sodium into the junctional cytosol to facilitate operation of NCX in the calcium influx mode such as calcium influx through reverse NCX [28]. Previous findings revealed that the bulk of calcium reloading of the SR during these repetitive calcium waves is mediated by the reversal of NCX linked to calcium uptake into the SR by SERCA [23]. In the current study, we found that the selective NCX blocker 3,4-DCB [29] completely abolished the PE-mediated contraction, suggesting these data are consistent with the involvement of NCX working in reverse mode (sodium out/calcium in) during PE-induced calcium entry. This also suggests that the activity of NCX largely modulates PE-mediated contraction. However, we do not know whether the role of NCX differs in the AMI group because the blocking effects of 3,4-DCB were too strong and we therefore could not distinguish this effect in the two groups. We also demonstrated involvement of the NCCE pathway on PE-induced contraction. However, there were no differences regarding the effect of the NCCE inhibitor RHC80267 on PE-induced contraction between the two groups. Furthermore, the relative contribution of the NCCE pathway towards the decreased PE-induced contraction in the AMI group remains unclear in the current study.
The current study indicates that the underlying mechanisms responsible for the change of vascular contractile or relaxing reactivity at the early stage of the post-infarction remodeling process may be associated with the enhanced NOS activity. However, it is still unclear which mechanisms are involved in the enhanced NOS activity after AMI, although some reports have demonstrated that eNOS may be activated by some mechanisms such as counter-humoral mechanisms [11] or superoxide [5,30]. In addition, recent study demonstrated that injury to the vessel wall is accompanied by a vascular smooth muscle cell (VSMC) phenotype switch from a contractile quiescent to a proliferative motile phenotype (synthetic phenotype), and alteration of many components of VSMC calcium signaling pathways. Specifically, this switch that culminates in a VSMC phenotype is characterized by loss of L-type VOCC expression and increased expression of T-type VOCCs and SOCCs. Therefore, future study should elucidate the underlying mechanisms responsible for the enhanced eNOS activity or involvement of the phenotype switch at the early period of the post-infarction remodeling process.
In this in vitro study, bath application using the relatively specific α1-AR agonist PE certainly did not mimic the release of NE, ATP, or vasoactive peptides at specialized sympathetic neuro-effector junctions. Furthermore, as the type and distribution of receptors and innervations varies with species and vascular beds, it may be expected that the physiological relevance of bath-applied α1-AR agonists will also vary. In addition, any clinical implications of PE-induced contraction in the current in vitro study must be tempered by the fact that a large conduit artery like the aorta was used in experiments. Even with these limitations, we believe that our results can provide valuable information concerning vascular hemodynamic changes such as acute coronary artery syndrome or AMI, and offer an effective strategy for the treatment of abnormal hemodynamic conditions.
In summary, we demonstrated a decreased sensitivity and efficiency of PE in rat aorta 3 days after AMI. We also showed a decreased sensitivity and maximal response for the VOCC inhibitor nifedipine under PE-mediated contraction after AMI, suggesting that VOCC-independent calcium entry mechanisms play a major role for PE-mediated contraction in rat aorta in the AMI group. Finally, we suggest that the enhanced CCE pathway via activation of SOCCs may be involved in these VOCC-independent calcium entry mechanisms in the AMI group. The main cause for the change of vascular contractile responses to PE may be associated with the enhanced eNOS activity during the post-infarction remodeling period. We expect that our results will be useful for the clinical management of hemodynamic parameters for cardiovascular intervention and coronary artery bypass grafting.

 XML Download
XML Download