

 PDF
PDF Citation
Citation Print
Print


Altered consciousness in patients is common in the intensive care unit (ICU). Fluctuations in the level of consciousness and/or behavioral disturbances can be caused by a number of factors including the medical conditions of the patient, substance intoxication, or medication side effects [1]. These patients require thorough neurological evaluation and subsequent appropriate management. However, in grave cases, rapid symptom management often holds priority over investigation of the underlying cause. Status epilepticus (SE) is a life-threatening condition in which the brain is in a state of persistent seizure. This condition is always considered a medical emergency and the mortality rate is quite high (7.6 to 43%), especially if treatment is not initiated quickly [2]. There are many causes of SE; metabolic abnormality and hyperammonemia as in our case are some of the rare etiologies. Clinical manifestations of hyperammonemia include hypotonia, seizures, emesis, and abnormal neurologic changes (including stupor). Ammonia levels above 200 µmol/L are reported to be associated with brain edema and herniation [3,4]. We report a case of sudden unexpected hyperammonemia that manifested with a sudden neurologic change and subsequent status epilepticus.
Case Report
A 39 year-old female patient with ovarian cancer (stage III) underwent TAH with BSO (total abdominal hysterectomy and bilateral salpingo-oophorectomy) at another hospital. During chemotherapy with paclitaxel and carboplatin (day 9, POD 16), she showed melena followed by hematochezia. Physical examination showed abdominal tenderness and rebound tenderness. Neutropenia was present in the blood test. She also had a high fever, tachypnea, hypotension, and cyanosis. She was diagnosed with neutropenic/septic shock due to ascending colon perforation and was transferred to our hospital. Emergent total colectomy was successfully conducted, and the patient was moved to the ICU. The patient was intubated, but alert and oriented before and after surgery.
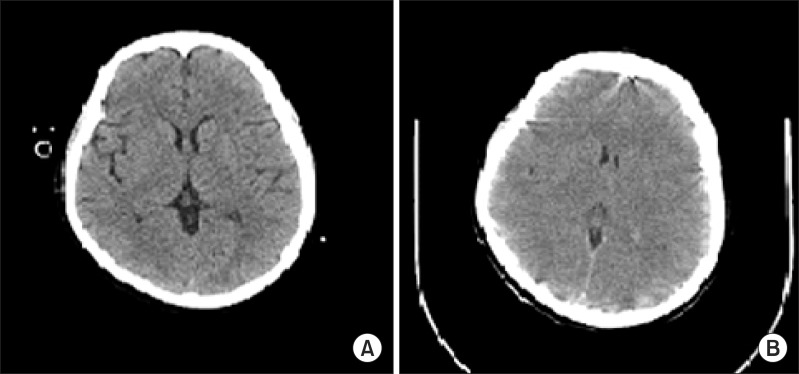
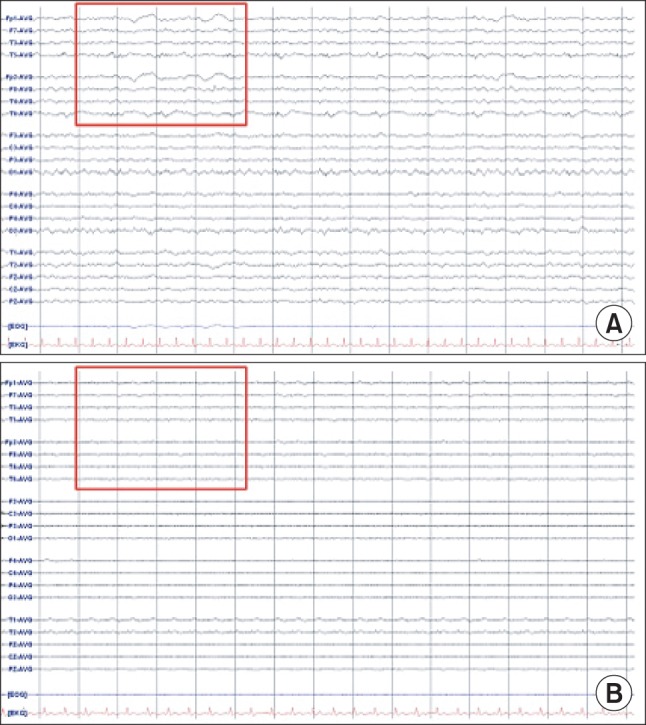
The patient experienced difficulty in weaning from mechanical ventilation due to severe tachypnea and labored breathing. On postoperative day 2, the patient was sedated with midazolam. Neutropenia did not improve in spite of granulocyte colony-stimulating factor (G-CSF) injection. Absolute neutrophil count (ANC) was less than 50/µl. Chemotherapy induced neutropenia and sepsis was suspected, thus G-CSF and antibiotics (Piperacillin/tazobactam) were administered. On postoperative day 5, midazolam was stopped. The patient was extubated but she complained of dyspnea and showed deteriorating levels of consciousness. After reintubation, brain CT and electroencephalography (EEG) were performed which showed no abnormal findings (Fig. 1A and 2A). Her neurologic status did not show improvement and on the next day, she suddenly developed partial seizure which progressed to generalized tonic-clonic (GTC) seizure. Seizure was controlled with midazolam, ativan, and phenytoin. Brain CT showed diffuse brain swelling (Fig. 1B) and the EEG test showed severe diffuse cerebral dysfunction (Fig. 2B). Laboratory tests indicated severe hyperammonemia (1,325 µg/dl). Arterial blood gas analysis showed respiratory alkalosis with metabolic alkalosis (pH: 7.604, PaCO2: 24.2 mmHg, HCO3-: 27.4 mmol/L), while electrolytes and liver panel were unremarkable. Continuous renal replacement therapy (CRRT) for the treatment of hyperammonemia was not effective (follow-up ammonia level was 1,356 µg/dl). Sodium benzoate was administered as a loading dose followed by a maintenance infusion. Mannitol was administered to attenuate the increased intracranial pressure. The patient showed recurrent episodes of GTC type seizure requiring management of status epilepticus. Midazolam and phenytoin were administered. In spite of medical treatment, further partial seizures occurred accompanied by desaturation and hypotension. The patient subsequently died on postoperative day 8.
Discussion
Fluctuating level of consciousness is common in ICU patients and it has been associated with increased mortality [5]. When serious neurological insults such as stroke or seizure affect a patient, it is often too late to attempt for a significant alteration in the outcome of the patient. Therefore, it seems best to identify the underlying risk factors and prevent/attenuate such critical conditions. However, given the multi-factorial and unpredictable nature of hyperammonemia, critical events can occur unexpectedly.
Clinical symptoms of hyperammonemia vary with patient age and ammonium level. Common symptoms of hyperammonemia include hypotonia, vomiting, lethargy, seizures, coma, ataxia, anorexia, abnormal behavior patterns, dysarthria, weakness, liver enlargement, and dementia.
Ammonia, along with inflammation, has been consistently shown to play a central role in hepatic encephalopathy [4]. Hepatic causes of hyperammonemia are well documented and include hepatotoxins, Reye syndrome, or carnitine deficiency [6]. However, hyperammonemia in the context of unremarkable liver function tests is a challenge for physicians-the differential diagnosis becomes more difficult.
Causes worth considering in patients with non-hepatic hyperammonemia include inborn errors of metabolism (defects of the urea cycle, fatty acid oxidation, organic acid disorders), circulatory shock with or without portosystemic shunt, hematological malignancies, urinary infection with urease-producing bacteria, anticonvulsants or chemotherapeutic drugs, and excessive amino acid load/increased catabolism [7]. Laboratory tests pertaining to enzyme deficiencies were not conducted in the present case because it was considered less worthwhile given the rapid clinical deterioration of the patient.
Hyperammonemia after chemotherapy has also been reported in the literature, mostly with hematological malignancies but also in colorectal cancer patients [8,9]. Hyperammonemic coma has been described in cases of postinduction chemotherapy for acute myeloid leukemia. It has also been described in patients with multiple myeloma in those receiving chemotherapy with aspraginase, 5-fluorouracil, and in those receiving rituximab. However, there have been no reports on chemotherapy agents (taxanes and platinum agents) used in the treatment of ovarian cancer, as the case in our patient. Surgical procedures or interventions reported to be associated with hyperammonemia include lung transplantation [10], bone marrow transplantation [11], and bariatric surgery [12]. Although the most common cause of hyperammonemia seems to be hepatic dysfunction, this is unlikely to be the cause in our patient given the normal liver function test results. We believe that the cause may have been infection, bleeding, chemotherapy or the sum of the factors listed.
The goal for the treatment of acute hyperammonemia should be rapid reduction of serum ammonia, regardless of the cause. Whenever possible, protein intake should be discontinued and hypercaloric glucose-based solution should be provided to enhance anabolism. Supportive treatment and correction of hydration, nutritional status, mineral (calcium, potassium), and electrolyte imbalances should be addressed. Nitrogen scavenger drugs, sodium benzoate and sodium phenylacetate, may be considered. Benzoate binds glycerine and forms hippurate while phenylacetate binds glutamine to form phenyl-acetylglutamine. Both are excreted in urine. When ammonia production exceeds removal, the role of dialysis becomes significant. On the other hand, dialysis may cause rebound increases in ammonia by removing nutrients from plasma and creating a catabolic state [13]. Orthotopic liver transplantation may be necessary in patients who display poor response to conservative management with refractory/recurrent hyperammonemia. One report showed that liver transplantation resulted in a 3-year survival of 93% and improved quality of life, including a normal-protein diet [14]. Given the high level of ammonia in our patient, CRRT and sodium benzoate were administered. Some advocate the use of hemodialysis which may have been more beneficial in our patient, but the substantial hemodynamic instability of our patient hindered its use.
Severe hyperammonemia is associated with high mortality and morbidity. When this is not primarily due to liver failure, prompt and early diagnosis followed by appropriate intervention is pivotal but challenging. With regard to our case, there were several possible causes of non-hepatic hyperammonemia that are unlikely when considered separately, but potentially possible when considered altogether. The possible causes that may have contributed to increased ammonia production include underlying infection, recent history of chemotherapy, GI bleeding, total parenteral nutrition [15]. In hindsight, serial levels of ammonia, including the baseline level, would have been helpful in managing the patient. Whether it would have altered the outcome remains uncertain. From the lessons of our case, we recommend that serum levels of ammonia should be included in the initial diagnostic work-up of patients with risk factors for hyperammonemia, especially if they show altered consciousness and convulsion/seizure in the ICU.
 XML Download
XML Download