

 PDF
PDF Citation
Citation Print
Print


Introduction
After Furchgott and Zawadzki [1] revealed in 1980 that an endothelium-derived relaxing factor (EDRF) is released from vascular endothelium, Palmer et al. [2] proved in 1987 that the EDRF was nitric oxide (NO). Whereas the conventional view attributed tissue injury process to ischemia itself, Bulkley [3] found that a variable proportion of the injury is caused by toxic oxygen metabolites that are generated from xanthine oxidase at the time of reperfusion. The metabolites were named reactive oxygen species (ROS) and included: superoxide radical (O2·), hydrogen peroxide (H2O2), and hydroxyl radical (OH·) [3]. Endothelium is particularly liable to damage by ROS [4], and endothelial dysfunction occurs after ischemia and reperfusion, characterized by a marked reduction in endothelium-dependent relaxation due to reduced release or action of EDRF [5]. Clinically, reperfusion injury may be possible by ischemic condition, including surgery, such as coronary artery bypass graft surgery, repair of aneurysm, and transplantation [6]. Fortunately, some intravenous and volatile anesthetics have antioxidant and free radical scavenging properties [7], and local anesthetics also attenuates cell injury induced by ischemicreperfusion [8].
Steroid is known to have neuroprotection effects in acute spinal-cord injury [9]. However, no study has yet shown that steroid has endothelial protection effect in the ischemia-reperfusion injury by ROS. In addition, dexamethasone to treat brain edema relate to the expression of vascular endothelial growth factor rather than the protection of brain-derived microvessel endothelial cells [10].
We studied the influence of methylprednisolone (MPD) and hydrocortisone (CRT) on ROS effects using the endothelium of rabbit abdominal aorta to examine if their anti-oxidizing effects can suppress or reduce the vascular endothelium injury by ROS.
Materials and Methods
Preparation of ring slices and recording
All experiments were conducted conforming to the regulations of the Laboratory Animal Committee. Auricular intravenous injection of heparin 600 IU/kg was carried out in the rabbits under sevoflurane inhalation anesthesia, and exsanguinations followed after 3 minutes by cleaving the carotid artery. The abdominal aortas of the rabbits (2-2.5 kg, Male, n = 27) were extirpated and 3-4 mm long ring slices were prepared by separating the lipid tissue and connective tissue, without engaging tension on them, in a petri dish containing Krebs-Henseleit solution (K-H solution: NaCl 120.0, NaHCO3 25.0, KCl 5.0, MgSO4 1.2, CaCl2 2.5, NaH2PO4 1.4, glucose 11.0 mM), and 95% oxygen and 5% carbon dioxide were insufflated. While maintaining the temperature at 37 ± 0.5℃, one end of the aorta slice was fixed in the tissue bath containing 5 ml of K-H solution. The opposite end was connected to a force displacement transducer (TSD 125®, Biopac Inc., USA), and the K-H solution was exchanged every 15 minutes during the 90-minute equilibration period. The resting stage tension was fixed at 2.0 g. The vascular smooth muscle tension was recorded using an amplifier (DA100C®, Biopac Inc., USA) with a data acquisition system (MP100®, Biopac Inc., USA) and a personal computer. Following the precontraction with norepinephrine (NE) 10-6 M, acetylcholine (ACh) 3 × 10-8, 10-7, 3 × 10-7, and 10-6 M were consecutively injected to observe the change of the aortic tone. Changes of the aortic tone by ACh injection before ROS exposure (control) and after ROS exposure (experimental) were compared.
Method of exposure to ROS
After obtaining the control group values by the consecutive injection of ACh, each reagent was pretreated, and following a necessary interval, electrolysis was carried out by applying an electric current (constant current, 15 mA) to the positive and negative electrodes in the K-H solution for 35 seconds to generate ROS and induce vascular endothelial injury. In the procedure, distance over 1 cm was maintained between the abdominal aorta ring slices and the positive and negative electrodes to avoid a direct stimulus of the electric field to the tissue. The K-H solution was exchanged, the precontrcaction by NE followed, and Ach was consecutively injected. Changes in the aortic tone were recorded as the experimental group values. The electrode was made of platinum (7.5 mm) ,and 15 mA of constant current generation was tested before the experiment.
Precontraction with NE and relaxation with ACh: control value
At the beginning of every experiment, precontraction was induced with NE (10-6 M), and once a stable tension plateau was established, Ach (3 × 10-8, 10-7, 3 × 10-7, and 10-6 M) were consecutively administered to observe the change of isometric tension (control value). We only used viable ring segments that showed more than 80% relaxation of aortic tension for the following procedures. We sustained 15 min of equilibrating period.
Effect of MPD and CRT pretreatment
After obtaining the control group values of the response to ACh before ROS exposure, the experimental group values were obtained by 15 minutes of experimental drug pretreatment, followed by exposure to the ROS for 35 seconds. Then, the K-H solution was exchanged, the precontrcaction by NE followed, and ACh was consecutively injected. The experimental groups included one group pretreated with MPD (10-4 M: n = 10, 3 × 10-4 M: n = 14, 10-3 M: n = 11, 3 × 10-3 M: n = 14) and the other group pretreated with CRT (10-4 M: n = 11, 3 × 10-4 M: n = 14, 10-3 M: n = 6, 3 × 10-3 M: n = 12), using concentrations of 10-4, 3 × 10-4, 10-3 and 3 × 10-3 M.
Effect of 3-amino-1,2,4-triazole (3AT) on vascular endothelial injury by ROS
The samples were pretreated with 5 mM 3AT, the catalase inhibitor, for 30 minutes [11]. After 3AT treatment for 15 minutes, 3 × 10-3 M of MPD (n = 7) and CRT (n = 11) were additionally added, and treatment was conducted for 15 minutes. Following 35 seconds of electrolysis to generate ROS, the K-H solution was exchanged, the precontrcaction by NE followed, and ACh was consecutively injected.
Effect of diethylthiocarbamate (DETCA) on vascular endothelial injury by ROS
The samples were pretreated with 0.5 mM DETCA, superoxide dismutase inhibitor, for 30 minutes. After DETCA treatment for 15 minutes, 3 × 10-3 M of MPD (n = 7) and CRT (n = 11) were additionally added, and treatment was conducted for 15 minutes. Following 35 seconds of electrolysis to generate ROS, the K-H solution was exchanged, the precontrcaction by NE followed, and Ach was consecutively injected.
Reagents
The reagents NE, ACh, 3AT, DETCA, were purchased from Sigma Co. (St. Louis, USA). The MPD was Solumedrol® (Pfizer Pharmacy, Korea), and the CRT was Solu-cortef® (Pfizer Pharmacy, Korea).
Statistical procedure
The degree of aortic tone relaxation by Ach was expressed as the percentage relaxation of the precontraction induced by NE (10-6 M). Data was presented as mean ± SE. Statistical analysis was performed using Prism 2.0™ (GraphPad software, San Diego, USA). N indicates the number of rabbit abdominal aortic rings. For the comparison of the results among the pretreated MPD and CRT concentrations, a one-way ANOVA was used. A paired t-test was used for the comparison of results before and after the ROS exposure, while an unpaired t-test was used for the comparison of the results between the 3AT or DETCA pretreated group and the non-pretreated group. All the post-hoc tests were carried out by the Dunnet test, defining statistical significance as P < 0.05.
Results
Vascular relaxation before ROS exposure (control)
Aorta ring segment contraction was induced by NE 10-6 M into a stable tension plateau. Cumulative ACh (3 × 10-8, 10-7, 3 × 10-7, and 10-6 M) administration following the plateau gave rise to aorta ring segment relaxation in a dose-dependent manner. The percentage of aortic tone relaxation by Ach was -93.3 ± 0.8%.
Vascular relaxation after vascular endothelial injury induction with ROS
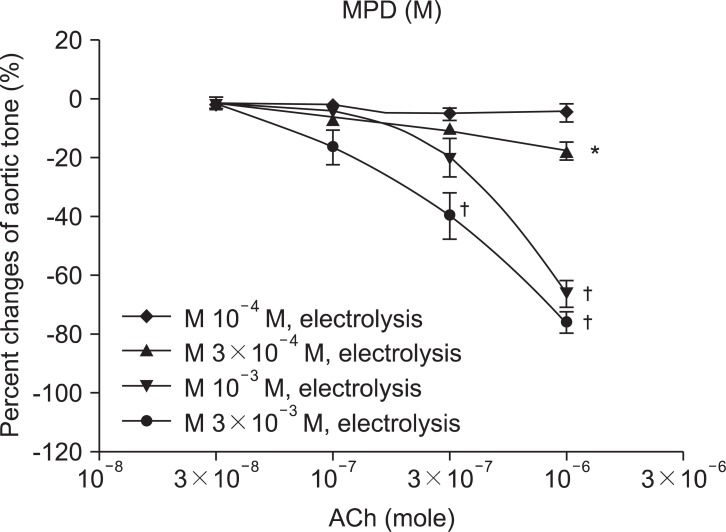
MPD 10-4, 3 × 10-4, 10-3, and 3 × 10-3 M groups enhanced relaxation by ACh 10-6 M in a dose-dependent manner. The percentage of aortic tone change by ACh 10-6 M was -4.6 ± 2.9, -17.6 ± 3.1, -66.0 ± 4.6, and -75.7 ± 3.6% in each, respectively. Comparing the MPD 10-4 M group with the MPD 3 × 10-4, 10-3, and 3 × 10-3 M groups, the percentage change of aortic tone was affected by MPD concentration with statistical significance (Fig. 1).
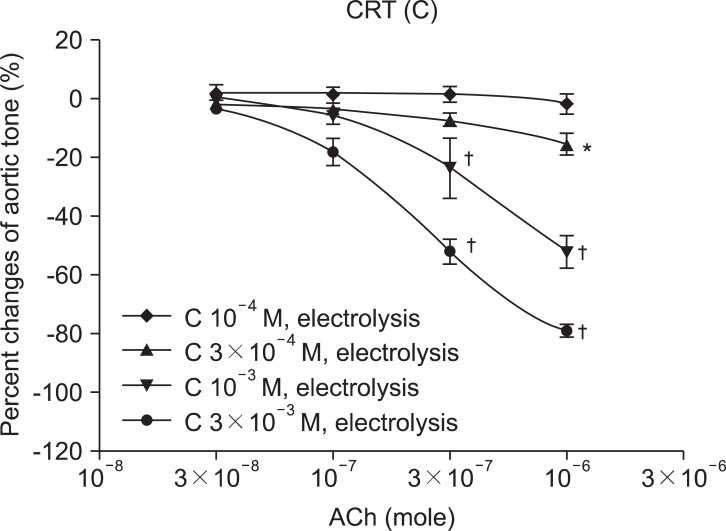
CRT 10-4, 3 × 10-4, 10-3, and 3 × 10-3 M groups enhanced relaxation by ACh 10-6 M in a dose-dependent manner. The percentage of aortic tone change by ACh 10-6 M was -2.0 ± 3.2, -16.0 ± 3.9, -52.0 ± 5.5, and -78.8 ± 2.2% in each, respectively. Comparing the CRT 10-4 M group with the CRT 3 × 10-4, 10-3, and 3 × 10-3 M groups, the percentage change of aortic tone was affected by CRT concentration with statistical significance (Fig. 2).
Effect of 3AT on vascular endothelial injury by ROS
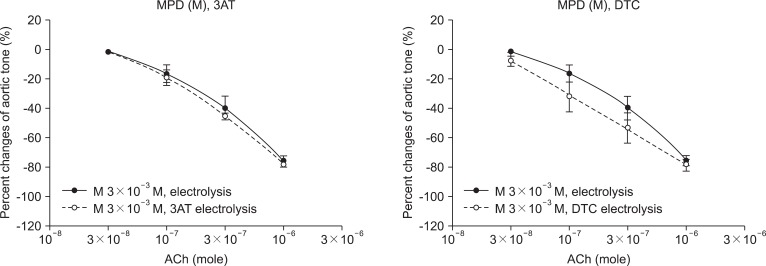
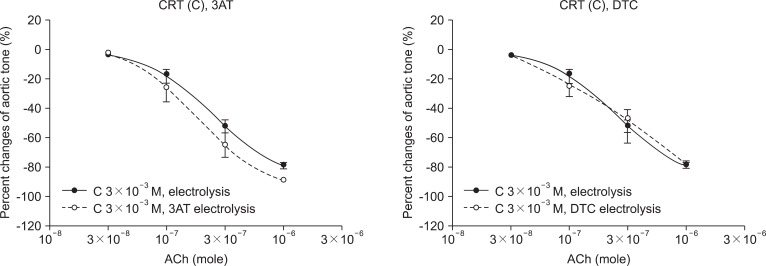
In the case of the MPD pretreated group (3 × 10-3 M), no significant change was found in the 3AT pretreated group (-77.2 ± 2.4%) compared with the unpretreated group (-86.0 ± 5.2%) (Fig. 3). As well in the case of the CRT pretreated group (3 × 10-3 M), no significant change was found in the 3AT pretreated group (-88.8 ± 0.8%) compared with the unpretreated group (-88.6 ± 1.9%) (Fig. 4).
Effect of DETCA on vascular endothelial injury by ROS
In the case of the MPD pretreated group (3 × 10-3 M), no significant change was found in the DETCA pretreated group (-78.7 ± 3.9%) compared with the unpretreated group (-89.2 ± 0.6%) (Fig. 3). As well in the case of the CRT pretreated group (3 × 10-3 M), no significant change was found in the DETCA pretreated group (-77.9 ± 2.2%) compared with the unpretreated group (-89.7 ± 1.7%) (Fig. 4).
Discussion
Reactive oxygen species (ROS) are normally generated at basal amounts, but ischemia or oxygen re-supply during reperfusion lead to abnormally high ROS levels which cannot be cleared by normal anti-oxidizing systems, causing tissue reperfusion injury [12]. In this process, an oxygen (O2) molecule generates the intermediate species, O2·, H2O2, and OH· and the intermediates O2· and OH· are important free radicals that cause peroxidation (LOO·) of intracellular lipids and tissue damage [13]. In particular, OH·, the most reactive ROS [1] is produced by the Fenton reaction (Fe2+ + HOOH → Fe3+ + OH· + OH-) and the Haber-Weiss reaction (O2·- + NO· → OONO- + H+ → OONOH → NOO· + OH·) [13]. ROS generated during reperfusion causes organ damage in the heart [14,15], brain [16,17], and blood vessels [18,19].
In 1980, Furchgott and Zawadzki [1] found an EDRF later shown to be NO [2]. Other vasoactive substances, including endothelium-derived hyperpolarizing factor and the endothelium-derived contracting factor, are released from the endothelium to regulate vascular tone. However, because the endothelium is composed of a cell monolayer, it is easily damaged by physical and chemical stimuli, including ROS during reperfusion. Inhalation anesthetics [12,15,20] and lidocaine, a local anesthetics, can prevent ischemia-reperfusion injury in the brain [13]. Steroid is known to have neouroprotection effects in acute spinal-cord injury [9]. However, no study has shown that steroid has endothelial protection effect in the ischemia-reperfusion injury by ROS. In addition, dexamethasone to treat brain edema relate to the expression of vascular endothelial growth factor rather than the protection of brain-derived microvessel endothelial cells [10].
We studied the influence of MPD and CRT on ROS effects using the endothelium of rabbit abdominal aorta to examine if their anti-oxidizing effects can suppress or reduce the vascular endothelium injury by ROS. In this study, both MPD and CRT dose-dependently protected endothelial-dependent relaxation by removing or anti-oxidizing ROS. In addition, their antioxidant effect was not disturbed by pretreatment of 3AT and DETCA.
We generated ROS by direct-electrolysis of a physiologic salt solution, the K-H solution [19,20]. It is known that ROS, such as O2·, H2O2, OH·, are generated during the physiologic salt solution electrolysis [21-24]. We did not add normal levels of ascorbic acid (1.1 × 10-4 M) into K-H solution to exclude its antioxidizing effect. In one study, using mouse chest aorta, it was reported that 5 minutes of electrolysis was required to induce injury by ROS [19]. However, 5 minutes of electrolysis with 15 mA constant current caused too much injury by ROS in the rabbit abdominal aorta, and the ROS generated by 35 seconds of electrolysis was sufficient for this study.
It was reported that vascular relaxation induced by ACh is caused by induced-release of NO in the vascular endothelium, and that ROS inactivates NO [25-27]. In addition, the ROS generated by ischemia or reperfusion causes the vascular endothelium injury [22,28]. In this experiment, after ROS was generated by 35 seconds of electrolysis, the K-H solution was replaced by a new solution, and the measurement was carried out following 15 minutes of the equilibrium period. As the measurement was carried out following the removal of ROS in this manner, blocking of the ACh vascular relaxation induction effect is assumed to be the result of the direct vascular endothelium injury by ROS, not the result of NO inactivation by ROS [26].
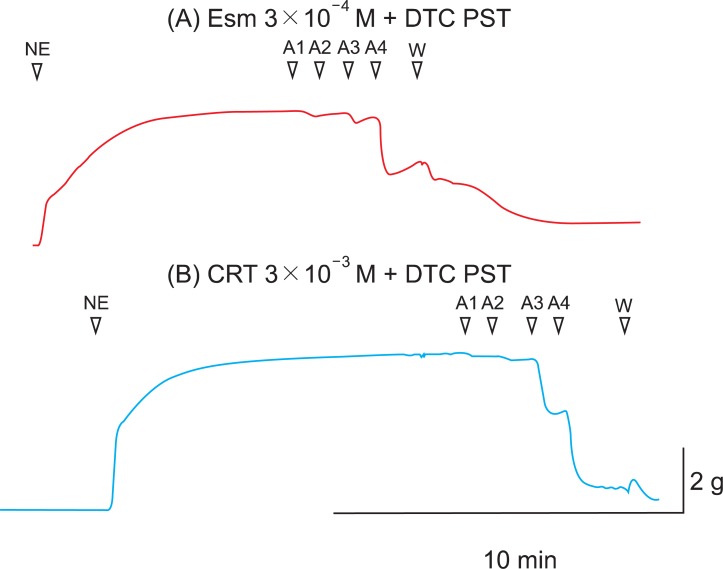
In this experiment, the optimal concentration of 3AT (5 mM) as catalase inhibitor was known from the previous study [11], but the optimal concentration of DETCA was not known. Before the main study, an earlier additional study was carried out to find the optimal concentration of DETCA that attenuates the superoxide scavenging effect. β-Blockers are strong antioxidants with the effects of scavenging lipid peroxyl radicals, and their antioxidant effects such as ROS scavenging are attenuated by DETCA [29,30]. In these experiments, among β-blockers, esmolol was used as an agent to find the optimal concentration of DETCA. Incremental doses of DETCA were pretreated (0.1, 0.2, 0.3, and 0.5 mM), and DETCA 0.5 mM interrupted the antioxidant effects of esmolol. Unlike the CRT group, significant attenuation of Ach-induced relaxation was found in the DETCA 0.5 mM pretreated esmolol group compared with the unpretreated group (Fig. 5).
In the study of the vascular relaxation after vascular endothelial injury induction with ROS, the MPD and CRT groups were relaxed by ACh in a dose-dependent manner. In MPD and CRT 3 × 10-3 M groups, with relatively high concentrations compared with MPD and CRP 10-4 and 3 × 10-4 groups, enhanced ACh-induced relaxation was observed. These findings suggest that MPD and CRT preserve the endothelium-dependent vasorelaxation against the attack of ROS, in a dose-related manner.
O2· is the first ROS which may lead to secondary radicals such as H2O2 and OH·. O2· is converted by superoxide dismutase to H2O2, which eventually becomes H2O by catalase. However, excess O2· is converted by the Fenton reaction and Haber-Weiss reaction to OH·, which is another pathway of ROS. In the study of the effects of 3AT and DETCA on vascular endothelial injury by ROS, the relaxation of the MPD and CRT 3 × 10-3 M groups were not disturbed by pretreatment of 3AT and DETCA. As such, the antioxidant effects of MPD and CRT are not attenuated by pretreatment of 3AT and DETCA and protect the endothelium of abdominal aorta. Endothelial protection mechanisms of MPD and CRT may not be H2O2 and O2· scavenging, but the scavenging of OH·, a remainder of ROS. However, to date, there is no specific inhibitor of OH·. Therefore, it is difficult to prove the mechanism of antioxidant effects of MPD and CRT with the manner of specific inhibitors such as 3AT and DETCA. This mechanism may be clarified through further studies with quantitative methods.
With the objective of this study to assess the influences of corticosteroids on the endothelium of rabbit abdominal aorta in ischemia and reperfusion, there are a few limitations. First, the clinically relevant concentration and relative potency of MPD and CRT could not be considered. The major difference between the two is that MPD has a 5 : 1 potency of glucocorticoid and <0.01 : 1 potency of mineralocorticoid compared to CRT. However, it was shown that there were dose-dependent protective effects of endothelium in two group. Therefore, it is believed that both MPD and CRT have antioxidant effects in ischemia and reperfusion. Second, clinical implication must be tempered by the fact that these results were obtained in a conduit-vessel-like aorta, because the diameter of true resistance vessels to regulate organ blood flow is 100-300 µm. Despite this, it was shown that corticosteroids preserved endothelium-dependent vasorelaxation against ROS.
In conclusion, it was tested whether ROS causes dysfunction in the rabbit aorta endothelium after MPD and CRT pretreatment. Pretreatment with MPD and CRT dose-dependently showed anti-oxidizing or ROS removing effects to protect endothelial-dependent relaxation in the abdominal aorta after ROS exposure. DETCA (superoxide dismutase inhibitor) and 3AT (catalase inhibitor) did not attenuate its protective effect. Further experiment to quantify the ROS is needed to investigate the protective mechanism of steroids.
 XML Download
XML Download