

 PDF
PDF Citation
Citation Print
Print


Introduction
Human neutrophils appear to be the major contributors to the first line of defense against microorganisms and critical effector cells in both innate and humoral immunity [1]. However, excessive activation of neutrophils may be deteriorative to the body, and even cause host tissue destruction in inflammatory diseases, such as rheumatoid arthritis, acute respiratory distress syndrome and septic shock [2]. Modulation of neutrophil function may provide a potential therapeutic strategy for these pathologic conditions.
Lipopolysaccharide (LPS), a gram-negative bacterial endotoxin, is thought to have a key role in the pathogenesis of sepsis and septic shock [3]. Interaction between LPS and Toll-like receptor 4 on neutrophil membrane initiates activation of neutrophils through a cascade of pathophysiological reactions [4]. The reactions include phosphorylation of mitogen activated protein kinases (MAPKs) and activation of nuclear transcription factor-kappa B (NF-κB) [5,6].
MAPKs are known to be involved in the signal transduction pathways of inflammation. Furthermore, MAPKs comprise three major subfamilies, p38 MAPK, extracelluar signal-regulated protein kinases 1 and 2 (ERK1/2) and c-Jun N-terminal kinase (JNK) [7]. NF-κB is a protein complex that controls DNA transcription. Activated NF-κB is translocated into the nucleus and the DNA/NF-κB complex then induces the expression of a wide variety of genes involved in inflammation, which results in a change of cell function [8].
It is important to modulate these pathways for the treatment of inflammatory diseases. Thus, numerous studies have been performed to find nontoxic molecules with anti-inflammatory activities.
Urinary trypsin inhibitor (UTI) is one of the Kunitz-type serine protease inhibitors found in human urine and blood [9]. It has been shown to inhibit a large number of proteases [10,11]. However, it was recently discovered that UTI is not just a protease inhibitor, but it has other physiological functions as a growth factor and a regulator of inflammatory response [12,13].
Anti-inflammatory effect of UTI seems to be due to the inhibition of leukocyte activation, but the mechanism for this has not yet been fully elucidated. Previous investigations have revealed this effect in LPS-induced inflammatory animal models [14,15]. In another series of studies, UTI was shown to inhibit the enhanced production of pro-inflammatory mediators such as interleukin (IL)-8, tumor necrosis factor-alpha (TNF-α) and thromboxane B2 in bronchial epithelial cells or monocytes [16-18]. However, there is no report on anti-inflammatory activity of UTI and its action mechanism in isolated human neutrophils.
This study was aimed to investigate the effect of UTI treatment on LPS-stimulated neutrophils involving production of inflammatory cytokines and to determine its intracellular signaling pathway.
Materials and Methods
Materials
Urinary trypsin inhibitor (Ulinastatin®) was generously gifted from Halim pharmaceutical (Seoul, Korea). Escherichia coli 055 : B5 endotoxin was purchased from Sigma-Aldrich (St. Louis, MO, USA). Immunoreactive TNF-α and IL-6 were quantified using commercially available enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA). RPMI 1,640/25 mM HEPES/L-glutamine was obtained from Mediatech (Herndon, VA), while FBS and penicillin/streptomycin were purchased from Mediatech. Bicinchoninic acid (BCA) protein assay reagent was obtained from Pierce (Rockford, IL, USA). Antibodies specific for phosphorylated (p)-ERK1/2, JNK and p38, as well as total ERK, JNK and p38 were purchased from Cell Signaling Technologies (Beverly, MA, USA).
Isolation of neutrophils
For isolation of human neutrophils, peripheral blood was obtained from healthy volunteers under a protocol approved by the institutional review board. None of the volunteers had a history of infection or allergy; none of them were smokers or undergoing immunosuppressive therapy. Neutrophils were isolated by plasma-Percoll gradients after dextran sedimentation of erythrocytes, as described previously [19]. Dextran was added to a final concentration of 6%, and erythrocytes were sedimentated under gravity for 45 minutes at room temperature. The erythrocyte-depleted supernatant containing leukocytes was centrifuged at 1,100 rpm for 6 minutes. The resulting leukocyte-enriched pellet was resuspended in platelet poor plasma (PPP). The leukocytes were then layered over a discontinuous gradient of percoll (2 ml of 42% and 2 ml of 51%) in a 15 ml polypropylene centrifugation tube. Percoll densities were created from stock percoll diluted with PPP. The percoll densities and layered cells were then centrifuged at 1,100 rpm for 10 minutes. Neutrophils were found at the 51-42% percoll layer interface. Moreover, when a few red blood cells (RBCs) stayed with neutrophils, we were able to use RBC lysing buffer (SIGMA, UK) for the lysis of RBCs. Neutrophils were removed from the new tube and washed twice with phosphate buffered saline (PBS). Cells were counted with a hemocytometer (Marienfeld, Germany) and resuspended at 5x106cells/ml with RPMI-1640 containing 5% of serum. Purity (Wright's stain) and viability (trypan blue exclusion) of the isolated cells were kept more than 97% and 95%, respectively.
Enzyme-linked immunosorbent assay (ELISA)
Neutrophils (5 × 106/ml) were incubated for 4 hours with varying concentrations of UTI (1, 10, 100, 1,000 and 10,000 U/ml) or LPS (100 ng/ml) alone or LPS plus UTI. Unstimulated neutrophils, exposed to neither UTI nor LPS, were also incubated as a control group. Concentrations of TNF-α and IL-6 in the culture media were determined using ELISA kits (R&D System, Minneapolis, MN, USA), according to the manufacturer's instructions and as described previously [20].
Western blot analysis
Neutrophils were incubated for 30 minutes with or without UTI (10,000 U/ml), or LPS (100 ng/ml) alone or with LPS plus UTI. Western blots were performed to detect the levels of phosphorylated and total p38, ERK1/2 and JNK, as previously described [20,21]. Neutrophils were lysed in ice-cold lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM Na2 EDTA, 1% Triton X-100, 1 mM EGTA, 1 mM Na3 vanadate, 2.5 mM Na pyrophosphate, 1 mM β-glycerophosphate, 1 mM PMSF, 1 µg/ml leupeptin, pH 7.5), and then resuspended and sonicated for 30 seconds. Debris from the lysed cell was pelleted by centrifugation at 14,000 rpm for 20 minutes. The supernatant was then removed and stored at -86℃. Protein concentration of each sample was assayed using the BCA protein assay kit (Pierce Chemical, Rockford, IL) standardized to BSA, according to the manufacturer's protocol. For the Western blot, 50 µg of protein was loaded and run on a 10% Tris-HCl SDS polyacrylamide gel. Protein was electrotransferred to a nitrocellulose membrane and then blocked with 5% non-fat dry milk, 20 mM TBS with 0.1% Tween. After blocking, the membrane was incubated overnight at 4℃ with a rabbit polyclonal specific primary antibody against p-p38, p-ERK1/2 and p-JNK, or at a dilution ratio of 1/1,000 in 1% BSA, followed by anti-rabbit or anti-rat Ig HRP-coupled secondary antibody at a dilution of 1/2,000 in 5% nonfat dry milk. After washing five times, bands were detected using ECL Western blotting detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ). The membranes were then stripped using stripping buffer (63 mM Tris-HCl, pH 6.8, 2% SDS, 100 mM 2-ME; from Bio-Rad, Hercules, CA), and reprobed with Abs specific for total ERK, JNK, or p38. Densitometry was performed by the chemiluminescence system and analysis software (Bio-Rad) to determine the ratio between phosphorylated and total kinases.
Electrophoretic Mobility Shift Assays (EMSAs)
Neutrophils were incubated for 1 hour with or without UTI (10,000 U/ml), or LPS (100 ng/ml) alone or with LPS plus UTI. EMSAs were performed, as previously described [20]. To obtain nuclear extracts from neutrophils, cells were resuspended in buffer A containing 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT, and the samples were incubated on ice for 20 minutes. After removing the cytoplasm from the nuclei by 15 passages through a 25-gauge needle, the nuclei were collected by centrifugation at 600 × g for 6 minutes at 4℃. The pellets were suspended in buffer C containing 20 mM HEPES, 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 25% glycerol, and 0.5 mM PMSF. After 30 minutes of incubation on ice, the suspension was centrifuged at 14,000 × g for 20 minutes at 4℃, and the supernatant was collected. Protein concentration in the supernatants was determined using BCA protein assay kits (Pierce Chemical, Rockford, IL). The nuclear extracts (5 µg) were incubated at room temperature for 20 minutes in 20 µl of reaction buffer containing 10 mM Tris-HCl (pH 7.5), 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, and 4% glycerol with a 32P end-labeled, double-stranded oligonucleotide probe specific for the κB site, 5'-AGTTGAGGGGACTTTCCCAGGC-3' (Geneka, Burlington, VT), and 1 µg of poly(dI-dC)·poly(dI-dC). In some experiments, unlabeled NF-κB or CREB oligonucleotide (Promega, Madison, WI) was added to the samples at 200-fold excess before adding the labeled probe. The sample was then incubated for 15 minutes on ice. The complexes were resolved on 5% polyacrylamide gels in Tris-HCl (pH 8.0)-borate-EDTA buffer at 10 V/cm. The dried gels were exposed to Kodak Biomax MS film (Eastman-Kodak, Rochester, NY) for 1-24 hours at -70℃.
Statistical analysis
Statistical analyses of the data were performed by two-way analysis of variance with repeated measures. The Scheffé test was used for multiple pair-wise comparisons when a significant difference was indicated through analysis of variance. The Kruskal-Wallis H test was used for group comparisons. A value of P < 0.05 was considered significant.
Results
UTI attenuates LPS-induced cytokine expression by neutrophils
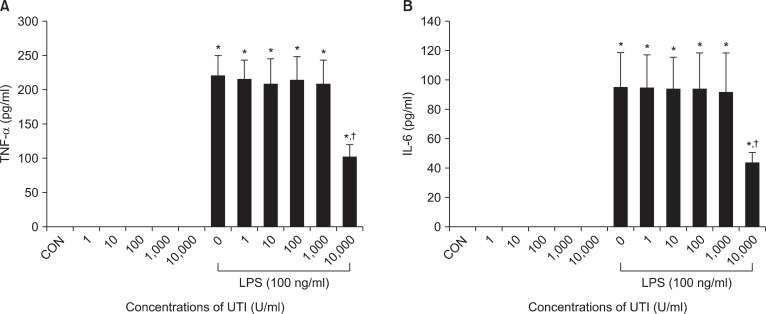
To assess the possible interactions between UTI and LPS on neutrophil activation, neutrophils were incubated with varying concentrations of UTI (1, 10, 100, 1,000 and 10,000 U/ml) plus LPS (100 ng/ml) or LPS alone, and protein levels for IL-6 and TNF-α were determined 4 h later. In these experiments, expression of both cytokines increased after stimulation of LPS. There was no effect of UTI alone over the range of concentrations examined. This LPS induced-expression in both cytokines was attenuated in the presence of UTI (10,000 U/ml) (Fig. 1).
The effect of UTI on LPS-induced activation of p38, ERK1/2, JNK, and nuclear translocation of NF-κB
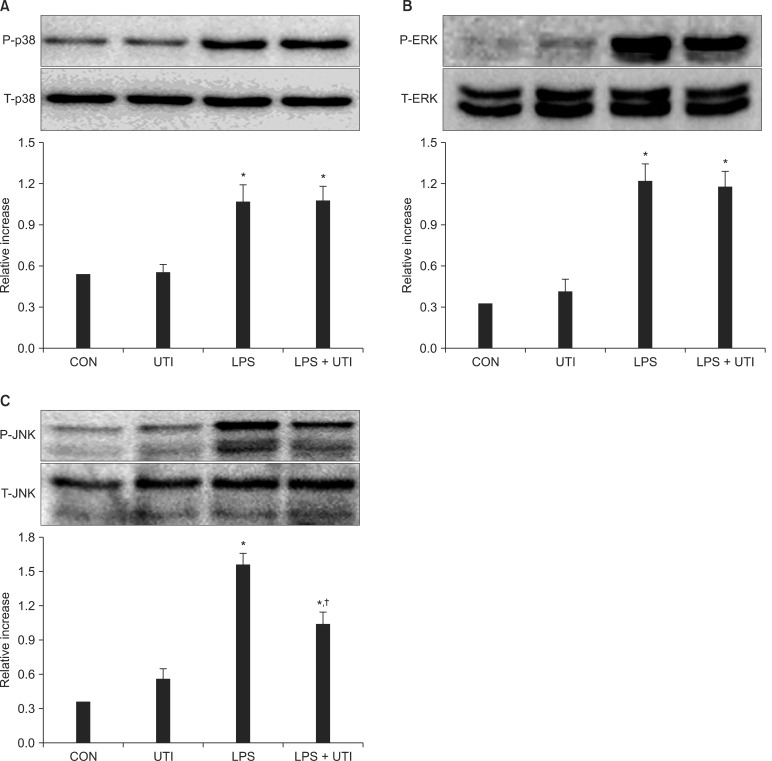
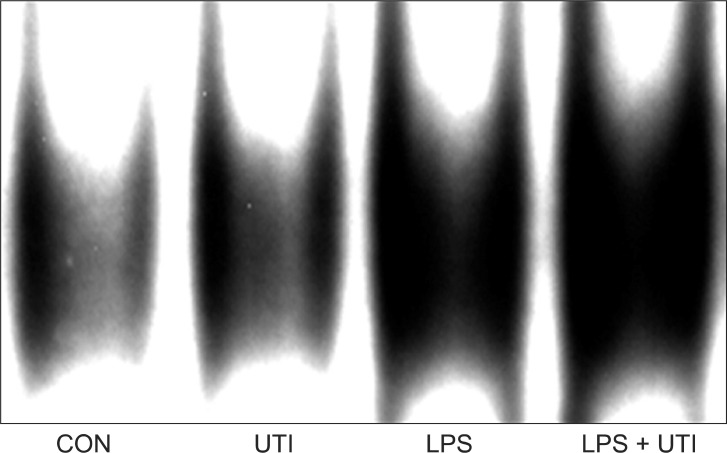
To examine the effects of UTI on LPS-induced activation of these kinase pathways and nuclear translocation of NF-κB, LPS-stimulated neutrophils were incubated with or without UTI (10,000 U/ml), and then the levels of active, phosphorylated forms of p38, ERK1/2, JNK, and nuclear levels of the transcriptional factor NF-κB were determined. Exposure of resting neutrophils to UTI alone had no effect on the activation of p38, ERK and JNK. As shown in Fig. 2, a culture of neutrophils with LPS resulted in the activation of p38, ERK1/2 and JNK. Addition of UTI to the neutrophils stimulated with LPS did not affect p38 and ERK1/2 activation, but did result in the attenuation of JNK phosphorylation. Activation of NF-κB induced by LPS was not affected by UTI (Fig. 3).
Discussion
This study was undertaken to examine the pharmacological effects of UTI on the production of inflammatory cytokines in human neutrophils. Moreover, we also investigated the effects of UTI on the activation of MAPKs and the transcription factor NF-κB.
The main finding of this study is that UTI decreased the expression of TNF-α and IL-6, and inhibited the activation of JNK, but not p38, ERK1/2 MAPKs and NF-κB in LPS-induced activated neutrophils.
Various kinds of mediators secreted from macrophages, monocytes and neutrophils, contribute to the initiation and progression of inflammatory diseases, such as ARDS and septic shock [22,23]. Among the different mediators, activated neutrophils are recognized to play crucial roles in the process of inflammation. When native neutrophils are stimulated with inflammatory stimuli, such as LPS, a rapid initiation of intracellular signal transduction takes place via sequential phosphorylation of kinases, which in turn leads to activation of MAPKs, followed by nuclear translocation of transcription factors like NF-κB and the ultimate synthesis of cytokine mRNA and proteins [6]. Through this signaling pathway, activated neutrophils release oxygen radicals, protease, leukotrienes, and inflammatory cytokines, such as IL-1β, IL-8, and TNF-α [23,24]. Inhibition of these inflammatory products is an important target in the treatment of inflammatory diseases.
UTI is a multivalent Kunitz type serine protease inhibitor that is found in human urine and blood. It is recognized to be degenerated from pre-α-/inter-α-trypsin inhibitors induced by neutrophil elastase during inflammation [25]. Recently, UTI has been used in Korea as a drug for patients with acute circulatory failure and pancreatitis. UTI mainly inhibits inflammatory proteases, including trypsin, α-chymotrypsin, plasmin, cathepsin G, and leukocyte elastase as well as proteases, in the coagulation cascade. As with other serine type protease inhibitors, UTI reportedly has anti-inflammatory properties other than blocking of the protease pathway in vitro. UTI inhibits the enhanced production of proinflammatory molecules such as IL-8 [16], TNF-α [17], thromboxane B2 [18] and prostaglandin H2 synthase-2 [26] induced by LPS in vitro. Similar with previous studies, the results of this study revealed that UTI attenuated the expression of TNF-α and IL-6 in LPS-induced neutrophils.
LPS activates MAPK cascades, such as p38, ERK1/2 and JNK, in humans [27]. In the current study, UTI treatment decreased the phosphorylation of JNK in response to LPS, but there were no detectable changes in p38 and ERK1/2 phosphorylation. These results show that anti-inflammatory activity of UTI is related to JNK-mediated pathways, but not p38 and ERK1/2 MAPK. However, Molor-Erdene et al.[28] reported that the mechanism of UTI inhibiting TNF-α production of monocytes was mediated by suppression of ERK1/2 activation. This divergence of intracellular signaling pathways may be due to the difference in cell lines.
The precise mechanism by which UTI inhibits LPS-induced phosphorylation of JNK remains unclear at present. Inanami et al. [29] reported that calcium ion released from intracellular stores is essential for phosphorylation of JNK in Chinese hamster lung fibroblasts, but not for p38 and ERK. Because UTI inhibits the increase in calcium ion influx and intracellular mobilization induced by LPS [30], it is likely that UTI inhibits LPS-induced JNK activation by inhibiting the increase of intracellular calcium ion.
NF-κB is a protein complex that controls the transcription of DNA. It is found in almost all animal cell types and is involved in the expression of many inflammatory genes and mediators responsible for the pathophysiology of inflammatory diseases [6]. In this study, however, nuclear translocation of NF-κB induced by LPS was not affected by UTI (10,000 U/ml). This result suggests that other transcription factors may be related to the anti-inflammatory effect of UTI, because the production of inflammatory mediators in neutrophils is regulated by various transcription factors, not only by NF-κB. Another hypothesis is that UTI inhibits either translation or secretion of inflammatory mediators. Further studies are required to determine the precise mechanism of inhibition of UTI on the production of inflammatory mediators in human neutrophils.
In conclusion, UTI decreased the expression of TNF-α and IL-6, and inhibited the activation of JNK in LPS-induced activated neutrophils. These results demonstrate that UTI can attenuate LPS-induced neutrophil responses and may play a partial role in the treatment of neutrophil-mediated inflammatory diseases.
 XML Download
XML Download