

 PDF
PDF Citation
Citation Print
Print


Introduction
Malignant hyperthermia (MH) is an inherited, pharmacogenetic skeletal muscle disorder involving the dysregulated myoplasmic Ca2+, hypercontracture, and hypermetabolism in response to an exposure to potent volatile anesthetics with/without depolarizing muscle relaxants, and can be triggered by exertional or heat stress without pharmacologic triggering agents in rare cases [1,2]. Susceptibility to malignant hyperthermia (MHS) is inherited as an autosomally dominant trait with variable expression and incomplete penetrance [1]. As almost all individuals with MHS appear normal without any pathologic signs, it is impossible to diagnose susceptibility without either the exposure to triggering agents or by specific diagnostic testing.
Although the estimated genetic prevalence of MHS is reported as up to 1 : 2,000-3,000 in individuals [3-5], MH episodes are very rare and modern anesthetic techniques, such as increasing use of non-triggering intravenous anesthetics and avoidance of succinylcholine are more likely to make it even rarer, leading to the potential risk of reduced awareness of anesthesiologists for MH.
When MH was first recognized as a complication of anesthesia, the mortality rate was 70-80% [6]. Nowadays, the mortality rate is estimated to be less than 5%, with early detection of MH episode, using capnography, prompt use of the drug dantrolene, and the introduction of diagnostic testing [7]. Even though the mortality rates of MH are low, according to a recent study by Larach et al. [8], the morbidity rate of MH is 34.8%. This high morbidity rate emphasizes the need for continuing education of anesthesiologists on the most effective way to diagnose and treat MH.
The purpose of this review is to summarize the clinically essential and important knowledge of MH and presents new developments in the field.
Go to : 
Epidemiology
Although the exact incidence of malignant hyperthermia is unknown, the incidence during general anesthesia is estimated to range from 1 : 5,000 to 1 : 50,000-100,000 in individuals [7,9]. However, the true number of individuals with MHS is likely to be much greater, as individuals with MHS may undergo anesthesia uneventfully several times before a clinical episode occurs and many cases of MH may go undetected because many individuals with MHS are never anesthetized, have short durations of exposure, or have mild, uncomplicated presentations that are never diagnosed. Generally, the estimated prevalence of MHS is thought to be approximately 1 in 50,000 or less. However, according to the results of recent molecular genetic studies, the estimated genetic prevalence may be up to 1 : 2,000-3,000 because MHS is inherited as an autosomally dominant trait [3-5].
MH has been reported in every country, and all races are susceptible [7]. MH occurs more frequently in males than females [7,8], and more commonly in children and young adults with the mean age of 18.3 years [10]. Although reported in the newborn, the earliest reaction confirmed by testing is six months of age [10]. Although the exact cause for male predominance of MH is unclear, suggested hypothesis includes differences in ionic channels for muscle contraction influenced by either sex hormones or native differences among genders, higher incidences false-positive diagnosis among males, as well as a difference in genetic inheritance [11-13]. In a prevalence and clinical outcome study from New York State from 2001 to 2005, the estimated prevalence of MH for males was 2.5-4.5 times the rate for females, and the median age at presentation was 22 years with 45% of MH cases occurring in those who were 19 years or younger [14].
The mortality rates of MH dramatically decreased from 70-80% to 10% after an introduction of dantrolene sodium for the treatment of MH, and recent mortality is estimated to be less than 5% with early detection of MH episode using capnography, prompt use of the drug dantrolene, and the introduction of diagnostic testing [6,7]. Although the mortality rates of MH are low, the morbidity rate of MH is much higher. According to the recent study of Larach et al.[8], the morbidity rate of MH is 34.8% and frequently reported complications are changes in the consciousness level/coma (9.8%), cardiac dysfunction (9.4%), pulmonary edema (8.4%), renal dysfunction (97.3%), disseminated intravascular coagulation (7.2%), and hepatic dysfunction (5.6%). This high morbidity rate emphasizes the need for continuing education of anesthesiologists on the most effective way to diagnose and treat MH.
Go to :
Pathophysiology
Malignant hyperthermia, a dominantly inherited pharmacogenetic skeletal muscle disorder characterized by skeletal muscle hypermetabolism, occurs in susceptible individuals following exposure to potent halogenated anesthetics or depolarizing muscle relaxants during anesthesia [7]. Although the exact mechanism by which triggering agents initiate MH has not been fully elucidated yet, numerous experimental evidences from various sources clearly indicate that underlying causes of this hypermetabolism are related to an uncontrolled release of calcium from skeletal muscle sarcoplasmic reticulum (SR) [1,2,15-18].
During excitation-contraction coupling, muscle membrane depolarization induces a conformational change in the dihydropyridine-sensitive L-type voltage-dependent calcium channel (DHPR). This conformational change leads to activation of the ryanodine receptor type 1 (RYR1) and the rapid release of Ca2+ from its stores in the SR. Released Ca2+ binds to troponin C, causing movement of tropomyosin away from the myosin-binding sites on the thin filament and triggering muscle contraction. Muscle contraction is terminated when Ca2+ is actively pumped back into the SR by the ATP-dependent calcium pump. This process is the fundamental excitation-contraction coupling (ECC) needed for normal skeletal muscle function [19,20].
In the early phase of MH, the muscle cells attempt to restore homeostasis by sequestering calcium through increasing in aerobic and anaerobic metabolism [7]. However, in malignant hyperthermia susceptible muscle, the uncontrolled release of calcium and the rise in myoplasmic calcium caused by the triggering agents overpowers the cellular capacity to reestablish homeostasis. This pathologically enhanced myoplasmic calcium rise eventually reaches the threshold levels for myofibrillar contraction, and results in sustained muscle contraction. The sustained muscle contraction produces a rapid depletion of adenosine triphosphate (ATP) with a concomitant increase in glucose metabolism, oxygen consumption, carbon dioxide production, and heat production. ATP stores become depleted, which progressively lead to the failure of membrane integrity with leakage of muscle cell contents (including electrolytes, myoglobin and various other sarcoplasmic proteins, like CK into the circulation [7].
The cause of dysfunctional calcium regulation in skeletal muscle with MHS lies, in most cases, in a defective RYR1 in SR, which is the footplate protein seated between the DHPR and SR [21-23]. Major defects in these two receptors (RYR1 and DHPR), as well as in other proteins, such as, triadin and FK 506 binding protein involved in the myoplasmic calcium regulation, are responsible for the functional changes of calcium regulation in MH. It is widely accepted that the gene of RYR1 (RYR1) is the major, though not exclusive, site for mutations associated with MHS [24,25]. Approximately 70% of families with MHS carry mutations in RYR1 [26-28].
Although mutations in RYR1 gene are undoubtedly important in the pathophysiology of MH, it is clear that not all families exhibit linkage to this gene. Other potential genetic loci have been identified, such as mutations in CACNA1S encodes the α1-subunit of DHPR [29,30].
Go to :
Clinical Presentation and Diagnosis
The diagnosis of MH is based on clinical presentations or laboratory testing. However, the clinical presentations and first signs of MH episode are highly variable, especially when some individuals exhibit only one or a few symptoms of varying intensity; it is not easy to diagnose MH with confidence [31,32]. Although the underlying causes for the variability of the clinical presentation are poorly understood, different potency, concentration and duration of exposure to the triggering inhalation anesthetics, as well as additional factors, such as temperature, age and genetic variability, may be related with the heterogeneous clinical presentations [31,33,34]. The variability in the order and time of onset of signs often makes the clinical diagnosis rather difficult [7,35,36].
The characteristic clinical signs of MH, during general anesthesia, are tachycardia, unexplained elevation of end-tidal carbon dioxide (ETCO2) concentration, muscle rigidity, acidosis, hyperthermia, and evidence of rhabdomyolysis. The most frequent and the earliest sign of MH is unexplained sinus tachycardia, together with unexplained elevation of end-tidal carbon dioxide. In a clinical analysis study of MH cases reported to the North American Malignant Hyperthermia Registry from 1987 to 2006 [8], frequent first signs of MH include hypercapnia (92%), sinus tachycardia (73%), and masseter muscle rigidity (27%). In 63.5% of cases, temperature abnormality (median maximum, 39.1℃) was the first to third sign. Whereas 78.6% presented both muscular abnormalities and respiratory acidosis, only 26.0% had metabolic acidosis.
The unexplained elevation of end-tidal carbon dioxide may be the earliest and the most common sign of MH episode in the intubated and mechanically ventilated patient [37]. Especially, when succinylcholine is used as the relaxing agent, abrupt rise of end-tidal carbon dioxide can be observed [38]. However, with a decline in the use of succinylcholine, a more gradual rise in CO2, rather than an abrupt rise are often noted, and it is possible to mask this rise by increasing the minute ventilation [39].
Sinus tachycardia is observed in almost all patients as one of the early signs of MH episode, and may be misinterpreted as an inadequate anesthetic depth.
Although the severe, rapidly developing hyperthermia is considered as a hallmark of MH from when malignant hyperthermia is described firstly in 1960 to nowadays, increasing body temperature is a relatively late sign of MH and is even absent sometimes [40,41]. Therefore, one should not wait for this sign to appear before making the diagnosis. When hyperthermia occurs, the rate of temperature increase may be as rapid as 1-2℃ every five minutes, up to greater than 44℃, and leads to a marked increase in oxygen consumption, carbon dioxide production, widespread vital organ dysfunction, and disseminated intravascular coagulation (DIC)[42]. The rate of temperature elevation is much more of a differential diagnostic and prognostic value than peak temperature, though higher maximum temperatures significantly increase the likelihood of MH complications [2,8]. For patient safety, a monitoring of core temperature is essentially recommended in all anesthetics expected to last ≥ 30 minutes [8,43].
Generalized muscular rigidity is a definitive sign of MH and reflects the Ca2+-dependent activation of muscle contraction accompanying activation of both aerobic and anaerobic metabolism. About 50-80% of the patients present generalized muscular rigidity [2]. If MH is suspected, peripheral muscular rigidity should be checked [41].
The change in arterial pressure is not usually marked in the early phase of MH. As MH episode progresses, late clinical signs, such as cyanosis, cardiac arrhythmias, mixed respiratory and metabolic acidosis, and various electrolyte imbalances arise.
Rhabdomyolysis is another clinical sign related to the destruction of skeletal muscle and is manifested by brown or colacolored urine. Acute renal failure may occur due to myoglobin precipitation in the renal tubules and close observation and laboratory monitoring for myoglobin should be instituted [44].
Disseminated intravascular coagulation (DIC) is a poor prognostic sign in MH. Larach et al. [45] found that DIC was associated with a 50-fold increased likelihood of cardiac arrest and an 89-fold likelihood of death.
Numerous conditions may resemble MH during anesthesia that present unspecific signs, such as tachycardia, hypercapnia, and hyperthermia, which may be confused with MH. These include surgical stress, inadequate anesthetic depth, sepsis, thyroid storm, pheochromocytoma, iatrogenic overheating, malfunction of anesthetic equipment, and etc. [7,41].
Larach et al. [46] had developed a clinical grading scale in order to assist in a clinical diagnosis of MH in 1994. The clinical grading scale is a scoring system that gives differential score to the various clinical manifestations of MH episode, such as muscle rigidity, muscle breakdown, respiratory acidosis, metabolic acidosis, temperature increase, cardiac involvement and family history. The scores are summed and categorized into six different groups to assess the likelihood of an intraoperative event representing an MH episode. The elements of the scoring system and interpretation are given in Table 1 and 2. This scoring system allows us some guidance in deciding whether a patient may be suffering from MH or not, without the use of specialized diagnostic testing. However, if data are partly or totally unavailable, which is common, the clinical grading scale lacks sensitivity to diagnose MH since evaluation using this scale depends on the availability of data [47,48].
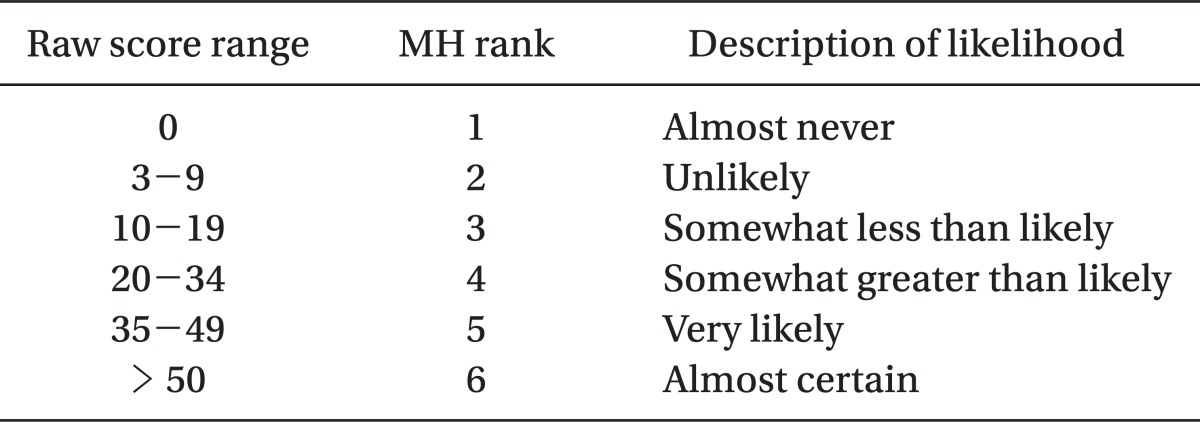

Table 2
Interpreting the Raw Score: MH Rank and Qualitative Likelihood (Adapted from Larach et al. [46])
![]()
Go to :
Laboratory Diagnostic Tests for MHS
As almost all individuals with MHS are asymptomatic during daily life, specific testing is needed to confirm or exclude MHS. In probands who have survived from a suspected MH episode, the confirmation of MHS diagnosis is very important because other family members may be affected by this dominantly inherited disorder. Also, the exclusion of MHS diagnosis in relatives of proband is equally important because MH is almost fatal if not rapidly diagnosed and treated. It would be a significant improvement for anesthesia risk management if we could reliably predict or confirm MHS [31,49].
The standard test for MHS diagnosis is the muscle contracture test, and recently, molecular genetic test is introduced.
The muscle contracture test
The most widely used and the most sensitive method for determining whether an individual is susceptible to MH is the muscle contracture test [32]. The muscle contracture test for diagnosis of MHS that involves exposing living muscle fiber bundles to halothane, and separately to caffeine according to a strict protocol, was developed based on the observations of Kalow et al. [50] and Ellis et al. [51] in 1970s.
After the processes of standardization (e.g., which muscle to biopsy, exposure regimens and diagnostic thresholds) to develop an accurate diagnostic test for MHS, this has been the "gold standard" for diagnosis of MHS. These processes of standardization led to the formation of two slightly different protocols, a European version (IVCT; the in vitro contracture test) by the European Malignant Hyperthermia Group (EMHG) and a North American version (CHCT; the caffeine-halothane contracture test) by the North American Malignant Hyperthermia Group (NAMHG) [52,53].
While similarities exist in performing and interpreting the results of tests, such as the criteria for muscle sampling, muscle biopsy extraction to testing time, and muscle viability assessments, there are significant differences between the two protocols [52,53]. When muscle is acquired anatomically, the NAMHG protocol may use any muscle site; whereas, the EMHG protocol limits muscle specimens from the vastus lateralis. The NAMHG protocol uses exposure to a single concentration of halothane (3%) with a range of diagnostic thresholds (0.2-0.7 g), while the EMHG protocol uses incremental exposure to halothane (0.5, 1, 2%) with a uniform diagnostic threshold (≥ 0.2 g). The NAMHG protocol considers a positive CHCT result if any one of the three bundles exposed to halothane or the three bundles exposed to caffeine exhibit a positive response (contracture of ≥ 0.7 g to 3% halothane or contracture of ≥ 0.3 g to 2.0 mMl/L caffeine), then the individual tested is judged to be MH susceptible (MHS); otherwise, he or she is considered normal (MHN) [54,55]. In contrast, the EMHG protocol requires a positive response (contracture of ≥ 0.2 g to ≤ 2% halothane) in one of the two bundles tested with halothane and a positive response (contracture of ≥ 0.2 g to ≤ 2.0 mmol/L caffeine) in one of the two bundles with caffeine to be diagnosed as MH susceptible (MHS) [56]. If the results of the halothane and caffeine tests are negative, then the individual is diagnosed as normal (MHN). If a positive response occurs only to halothane or to caffeine, the test result is labeled as MH equivocal (MHE), although the individual is regarded clinically as MHS. This individual is further subdivided into MHE (h) and MHE (c), depending on whether the positive response was to halothane or to caffeine, respectively [52]. Currently, North American testing centers use a lower threshold for MHS, referred to as a clinical diagnosis (contracture ≥ 0.5 g to 3% halothane, and a contracture ≥ 0.3 g to 2 mM caffeine), to include as many susceptible patients as possible with the possibility of misdiagnosing a patient as MHS when they might be MHN. When individuals are going to be used in genetic studies, a higher threshold (halothane contracture ≥ 0.7 g) for diagnosis of MHS is used. As such, only patients truly susceptible will be included in research [57].
Like any tests used in clinical medicine, muscle contracture testing has limits on its sensitivity and specificity. Because MH episode is potentially fatal, the thresholds for diagnostic testing require a high degree of sensitivity and an acceptable degree of specificity. This means that anesthesiologists are willing to accept false-positive responses to avoid false-negative ones because the consequences of a false-negative diagnosis might be disastrous. Consequently, the diagnostic thresholds were established to minimize the possibility of false-negative results.
The EMHG protocol may reduce the possibility of false positive and negative results, when compared to that of the NAMHG protocol; however, obtained results were similar, overall [58]. Despite the use of low cutoff for interpreting and risking higher false positive in the EMHG protocol, the IVCT has a 99% of sensitivity and a 93.6% of specificity, while the NAMHG protocol has a sensitivity and a specificity of 97-98% and 78-80%, respectively [55,56]. Thus, there are a 1% chance of misdiagnosing a MH-susceptible individual as MHN, and a 6% chance of misdiagnosing a individual without susceptibility as MHS in the EMHG protocol; whereas there are a 2-3% chance that test will incorrectly mislabel a MH-susceptible individual as MHN, and a 20% chance that a individual without susceptibility will be labeled as MHS in the NAMHG protocol.
Although the decision to proceed with muscle contracture test depends on numerous factors, including the features of the suspected MH event, family history, patients' willingness, etc, in general, muscle contracture test should be performed on individuals with a clinical history of MH or close relatives of known MH susceptible individuals to exclude MHS [32].
Even though muscle contracture test still regarded as the gold standard for the diagnosis of MHS, this test is invasive, expensive, and can be performed only at specialized centers. Therefore, the search for a less invasive method than IVCT/CHCT to diagnose MHS has been ongoing for many years, and molecular genetic testing based on advances in molecular genetics and cellular physiology has been introduced as an attractive alternative [32,59].
The molecular genetic testing
Although molecular genetic testing for MHS has been introduced relatively recently, it is used as a diagnostic test for MHS, in limited capacity, in Europe and North America [60-62].
Molecular genetic testing was first suggested as a method of MHS diagnosis in 1990, when a mutation within RYR1 encoding the skeletal muscle calcium release channel was identified [63]. Since then, it was identified that about 70% of MHS have been linked to RYR1 with over 300 mutations associated with MHS [28,61,64]. With the identification of causative mutations in RYR1, and the rapid developments in molecular genetics technology, molecular genetic testing is expected to be a screening test for MHS diagnosis. However, it is not realized at this time because of the metabolic complexity, discordance between phenotype and genotype, and genetic heterogeneity of MHS [65-68].
At present, despite more than 300 MHS associated mutations in RYR1 have been identified, only 30 functionally confirmed causative point mutations have been approved as diagnostic mutations for MHS by EMHG, and 29 have been formally accepted as causative mutations by the MAMHG [61]. This means that these only 30 causative mutations can be used for screening of MHS diagnosis. The most important limitation of screening of MHS, using molecular genetic test at this time, is that the absence of causative RYR1 mutations itself cannot rule out MHS without muscle contracture test. Therefore, molecular genetic test for MHS diagnosis should always be used in selected and genetically characterized families, as well as under the guidelines for molecular genetic test recommended by the EMHG [60,62].
When MHS is confirmed in a proband or appropriate family member by muscle contracture test and a causative mutation is identified, then molecular genetic test becomes valuable for other members of the family, as other members of the family can be diagnosed as MHS by relatively a simple test for the presence of the same familial mutation, without muscle contracture test. Mutation-positive members would be regarded as MHS without muscle contracture test. However, individuals in whom the familial mutation was not identified must undergo muscle contracture test, in order to confirm or exclude MHS [60,62].
Molecular genetic test for MHS diagnosis has some limitations, including the relatively low sensitivity (25%) and the considerable amount of interindividual and intraindividual variability in phenotypic expression in individuals with MHS [32,69]. Ongoing investigations will unveil further causative mutations, and thereby improve the usefulness and efficacy of molecular genetic testing in the future.
Go to :
Management of MH Episode
Early recognition of the early signs of MH episode and immediate starting of appropriate treatment is essential for successful treatment of MH as the prognosis is poorer if late signs appear. Although the early signs of MH episode are nonspecific, anesthesiologists should be familiar with the nature of the condition and clinical signs of MH episode, and maintain awareness during general anesthesia in order to diagnose rapidly. Table 3 lists the clinical signs associated with MH. Because the clinical presentation of MH episode highly varies, treatment should be modified accordingly [70].

Once MH episode is suspected, all triggering agents should be discontinued immediately and the vaporizer for the volatile anesthetic should be removed from the anesthesia machine. If the surgical procedure must continue after discontinuation of triggering agents, anesthesia should be maintained using non-triggering agents, including opioids, sedatives, and non-depolarizing muscle relaxants.
It is imperative to call additional experienced help immediately because the coordinated team approach will lead to the most efficient implementation of the recommended interventions.
The patient should be hyperventilated with 100% oxygen (>10 L/min) through a non-rebreathing circuit at 2-3 times the normal minute ventilation to correct hypoxemia and respiratory acidosis.
The only specific drug to manage MH is dantrolene sodium, a specific ryanodine receptor antagonist that inhibits the pathologically increased calcium release from the sarcoplasmic reticulum in the affected individuals [71,72]. The recommended initial dosage is 2.5 mg/kg administered intravenously every 5 minutes, up to the total of 10 mg/kg until the hypermetabolism is overcome and clinical signs subside. In some cases, the dose may be as much as 20 mg/kg. If more than 20 mg/kg of dantrolene is not successful, the diagnosis of MH should be reconsidered. After the acute crisis has been controlled, dantrolene 1 mg/kg every 4 to 6 hours or alternatively 0.25 mg/kg/hr continuous infusion for 24 hours is recommended. The prognosis is markedly influenced by the time between the onset of symptoms and the administration of dantrolene at a starting dose of 2.5 mg/kg [2,70]. Although dantrolene sodium has no direct effect on actin/myosin binding, and no effect on the neuromuscular junction [73], dantrolene sodium may cause significant muscle weakness in patients with pre-existing neuromuscular disease [7]. Therefore, it should be used with extreme caution in those patients. Adverse effects, associated with dantrolene sodium, include muscle weakness, a sense of respiratory inadequacy, fatigue, dizziness, blurred vision, nausea, and thrombophlebitis [71].
Hyperthermia, a late clinical sign, should be treated with a variety of cooling techniques, including the use of cold 0.9% sodium chloride intravenous fluids, lavage of the stomach, bladder, rectal, or open cavity, and placement of ice packs on the neck, axilla, and groin. During the application of the cooling maneuver, core temperature should be monitored at the appropriate monitoring sites, including distal esophagus, nasopharynx, tympanic membrane, rectum, bladder, or axilla, as the patient should be cooled to no less than 38℃ to avoid any additional problems.
Hyperkalemia may be life-threatening and precipitate cardiac arrhythmias; therefore, hyperkalemia should be aggressively treated with insulin, dextrose, sodium bicarbonate, and calcium.
The mixed respiratory and metabolic acidosis should be treated with hyperventilation at 2 to 3 times the predicted minute ventilation with 100% oxygen, and intravenous administration of 1-4 mEq/kg sodium bicarbonate. Repeated administration of sodium bicarbonate might be necessary because of a continuous efflux of lactate from cells as lactate only crosses through cell membranes slowly.
Initial management of cardiac arrhythmias includes treatment of the underlying acidosis and hyperkalemia as most arrhythmias during MH episode usually occur due to hyperkalemia and acidosis. If arrhythmias do not respond to the initial management, antiarrhythmic therapy using lidocaine, amiodarone and β-adrenoceptor blocking agents should be initiated. For the control of cardiac arrhythmias, calcium channel blocker (e.g., verapamil, diltiazem) should not be used as these drugs interact with dantrolene, and can lead to life-threatening hyperkalemia, myocardial depression and ultimately cardiac arrest [7,72].
As myoglobinemia and myoglobinuria, due to acute rhabdomyolysis, increase the risk of kidney damage, urine output should be maintained at greater than 2 ml/kg/hr with more aggressive hydration, urine alkalinization, and diuretics [74].
In addition to the monitoring devices already in place (temperature probe, electrocardiogram, and arterial oxygen saturation), additional monitoring devices, such as an arterial cannula, a central venous catheter, as well as a urinary catheter in order to monitor renal function, should be used.
Laboratory testing should be performed immediately at the onset of an episode and periodically thereafter. Laboratory monitoring parameters include arterial and venous blood gases, electrolytes, coagulation profile, myoglobin and creatine kinase (CK). Arterial blood gases, serum electrolytes, and blood sugar should be checked every 15 minutes until the clinical signs of MH subside. The concentration of CK should be measured every 6-8 hours, until the value is normalized as CK is a guide to the status of muscle destruction, and will peak at 14-20 hours after muscle damage occurs [41].
After stabilization, the patient should be closely observed in ICU for at least 36-48 hours because MH recurs in about 25% of patients within 48 hours of a treated episode [7,75]. Dantrolene sodium at a dose of 1 mg/kg should be continued every 4 to 6 hours intravenously during this period, depending on the clinical and laboratory parameters.
Go to :
MHS and Association with Idiopathic HyperCKemia and Other Neuromuscular Disorders
Idiopathic hyperCKemia
Idiopathic hyperCKemia (IH) is a persistently elevated serum concentration of creatine kinase (CK) i.e., at least 3 serum CK levels more than twice normal over at least 3 months, without weakness or other significant neuromuscular symptoms [76,77]. Persistently elevated serum concentration of creatine kinase (CK) usually accompanies muscle weakness in patients with myopathies. However, it may also be found in individuals with a normal neurological examination, possibly due to subclinical or preclinical neuromuscular disorders, dystrophinopathy carrier state, hypothyroidism, hypoparathyroidism, alcoholism, or intake of statins and other drugs, and this condition is labeled asymptomatic hyperCKemia [78-80]. When the cause is not found, even after extensive investigations, the condition is defined as IH. Capasso et al. [80] studied a series of individuals with IH and found IH is familial in 46% of cases and familial IH is a benign genetically heterogeneous condition that is autosomal-dominant in at least 60% of cases, with higher penetrance in men.
Individuals with IH have long been suspected to have MHS and various studies have concluded IH to be a weak predictor of MHS [81-83]. Weglinski et al. [84] studied 49 asymptomatic individuals with IH and reported 24/40 (49%), while in a more recent investigation of 37 Italian subjects with IH, muscle contracture test detected one out of 37 individuals to be MHS (2.7%) [85]. This discrepancy, between the results of the investigation, may be caused by different inclusion criteria of the investigation [35].
Although increased serum levels of CK are a valid reason to screen for underlying neuromuscular disorders, and to suspect MHS [86], no certain correlation has been reported between serum levels of CK and MHS [79,85,86]. Meanwhile, if serum level of CK is increased in an individual who has a first-degree relative proven MHS, then the individual can be considered to be susceptible [87]. Therefore, individuals with IH, who belonged to a family with a history of MH, could be susceptible to MH but IH itself can be used as a predictor for MHS.
Neuromuscular disorders associated with MHS
Although individuals with certain neuromuscular disorders have long been suspected to have MHS, the risk of MH or MH-like reactions in patients with known neuromuscular disorders is not easy to determine. Because a number of case reports and small series have discussed patients with these disorders, who developed one or more of the following clinical symptoms: rigidity, increased temperature, arrhythmia, rhabdomyolysis, and hyperkalemia, during or after anesthesia, the anesthesiologist is confronted with the question of whether there is a true association between these disorders and MHS. However, it is important to recognize an association between these disorders and MHS, as the different underlying pathophysiological mechanism is to require different treatment and have different implications for future anesthetic management of the patient and their family.
The neuromuscular disorders that have a strong evidence for close association with MHS include central core disease (CCD), multi-minicore disease (MmCD) with RYR1 mutation and King Denborough syndrome [88-90].
Central Core Disease (CCD) is a rare hereditary myopathy with autosomal dominant inheritance, which is characterized clinically by muscle weakness of variable degree and histological by a predominance of type I fibers, containing clearly defined areas (central cores) lacking oxidative enzyme activity [91]. Individuals with CCD have been suspected to have MHS early, because individuals with MHS may have central cores on muscle biopsy [89], and individuals with CCD may be prone to MH episodes [92,93]. In the majority of cases, mutations associated with CCD are found mainly in RYR1 gene [26,94]. Among the reported mutations in RYR1 associated with MHS or CCD, more than 150 mutations are associated with MHS, approximately 100 mutations predispose to CCD, and more than 20 mutations appear to be associated with both MHS and CCD[1,64]. Therefore, Individuals with CCD should be considered as MHS and should be avoided triggering agents.
Multi-minicore disease (MmCD) is a recessively inherited congenital myopathy, characterized clinically by neonatal hypotonia, delayed motor development, and axial muscle weakness, which leads to the development of scoliosis and significant respiratory involvement, and histological by multifocal, well-circumscribed areas (multi cores) with reduction of oxidative staining and low myofibrillar ATPase [95]. It is generally accepted that MmCD is associated with mutations in two genes, SEPN1 and RYR1 [96,97]. Even though the association with MHS is not well documented, clinical MH episodes have been recognized in few cases with MmCD [98,99].
King Denborough syndrome is a rare congenital myopathy associated with MHS, characterized by skeletal abnormalities, such as palmar simian line, pectus excavatum, winging of the scapulae, lumbar lordosis and mild thoracic scoliosis and dysmorphic features with characteristic facial appearance, such as dysmorphic facies, ptosis, down-slanting palpebral fissures, hypertelorism, epicanthic folds, low-set ears, malar hypoplasia, and micrognathia [7,100]. Inheritance of King-Denborough syndrome is unclear. Approximately one half of individuals with King-Denborough syndrome demonstrate the baseline elevated serum CK levels [101]. Recently, RYR1 mutation associated with King-Denborough syndrome are identified and reported [102]. Individuals with King-Denborough syndrome also should be considered as MHS and should not receive triggering agents.
Go to :
Current Status of Malignant Hyperthermia in Korea
In recent years, RYR1 mutations related MHS have been identified and reported in Korea [99,103,104]. However, the current state of MH in Korea is far from optimistic, in terms of underestimation of the incidence, poor diagnostic tests for MHS and limited availability of dantrolene sodium.
Because there is no national based MH database and case reports published on journals have been the exclusive resource for estimating the incidence of MH, the incidence of MH has long been underestimated in Korea. Only 27 cases have been reported during the year 1971-2007 [105]. The estimation of incidence based on the reported cases is underestimated, since the case reports published on journals only represent a part of the real number of MH cases. To estimate incidence with confidence, we should try to establish a national based MH database.
Although RYR1 mutations related MHS are discovered and reported in Korea, patients with MHS are mainly diagnosed on the basis of clinical manifestations without specific laboratory tests to confirm MHS; since there is no laboratory facility to confirm MHS who experienced MH episode during anesthesia.
Lastly, dantrolene sodium, the only specific drug for MH episode, can be available in Korea since several years ago. However, dantrolene sodium is still limitedly available, because dantrolene sodium is only supplied by KODC (Korean Orphan Drug Center).
The systemic efforts to improve the situations described above are required for the patients' safety in Korea.
Go to :
 XML Download
XML Download