

 PDF
PDF Citation
Citation Print
Print


Introduction
Dexmedetomidine is widely used during the perioperative period because of its anxiolytic, sedative, analgesic, and sympatholytic effects [1]. The selectivity ratio (α2/α1) of dexmedetomidine as a highly selective α2-adrenoceptor agonist is approximately eight times higher than that of clonidine [2].
Intravenous administration of dexmedetomidine induces initial, transient hypertension due to vasoconstriction caused by direct stimulation of the α2-adrenoceptor on vascular smooth muscle [3,4]. Vascular smooth muscle contraction is controlled by calcium-dependent and calcium-sensitization mechanisms [5]. Calcium-dependent smooth muscle contraction is associated with intracellular free calcium concentrations that are due to either calcium influx from the extracellular space or calcium release from the sarcoplasmic reticulum [5]. α2-Adrenoceptors are subdivided into α2A, α2B, and α2C subtypes [6]. Dexmedetomidine is a full agonist of the α2B-adrenoceptor and a partial agonist of the α2A-adrenoceptor, both of which are mainly involved in arterial contraction [6,7]. Dexmedetomidine-induced contraction involves calcium sensitization mediated by Rho kinase, protein kinase C, and phosphoinositide 3-kinase [8]. However, the calcium-dependent mechanism involved in dexmedetomidine-induced contraction remains unknown. Therefore, the goal of this in vitro study is to investigate the calcium-dependent mechanism underlying dexmedetomidine-induced contraction of isolated endothelium-denuded rat aorta.
Materials and Methods
All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee.
Preparation of aortic rings for tension measurement
Male Sprague-Dawley rats (n = 49), weighing 250-350 g each, were anesthetized by intraperitoneal administration of pentobarbital sodium (50 mg/kg). The descending thoracic aorta was dissected free of surrounding connective tissue and fat, and removed under microscopic guidance in a Krebs solution of the following composition: 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.4 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose. The aorta was then cut into 2.5-mm rings, suspended on Grass isometric transducers (FT-03, Grass Instrument, Quincy, MA, USA) under a 3.0-g resting tension in a 10-ml Krebs bath at 37℃, and aerated continuously with 95% O2 and 5% CO2 to maintain pH values within the range of 7.35-7.45. The rings were equilibrated at a 3.0-g resting tension for 120 min, changing the bathing solution every 30 min. The endothelium was removed from the aortic rings by insertion of a 25-gauge needle into the lumen of the rings and gently rubbing the ring for a few seconds. Once phenylephrine (10-8 M)-induced contractions were stabilized, endothelial denudation was confirmed by observation of less than 15% relaxation in response to acetylcholine (10-5 M). The contractile responses induced by isotonic KCl (30 mM) were measured for all aortic rings and used as a reference value (100%). An isotonic 30 mM KCl solution was prepared by replacing the NaCl in the Krebs solution with an equimolar amount of KCl. After washing KCl from the organ bath and returning the isometric tension to baseline, a cumulative concentration-response curve for dexmedetomidine was obtained as described below. Each ring was used for only one concentration-response curve induced by dexmedetomidine. The cyclooxygenase inhibitor indomethacin (10-5 M) and the nitric oxide synthase inhibitor NW-nitro-L-arginine methyl ester (L-NAME, 10-5 M) were also included in the Krebs solution to prevent the release of endogenous prostaglandins and nitric oxide, respectively, from any residual endothelium [8-10].
Experimental protocol
In the first set of experiments, we examined the effects of the α2-adrenoceptor inhibitor rauwolscine (3 × 10-6 and 10-5 M); the voltage-operated calcium channel blocker verapamil (5 × 10-7, 10-6 and 5 × 10-5 M); the purported inositol 1,4,5-trisphosphate (IP3) receptor blocker 2-aminoethoxydiphenylborate (2-APB, 5 × 10-6, 10-5 and 5 × 10-5 M); the store-operated calcium channel blocker gadolinium chloride hexahydrate (Gd3+; 5 × 10-6 M); and the phospholipase C inhibitor U-73122 (10-6 and 3 × 10-6 M) on cumulative dexmedetomidine concentration (10-9 to 10-6 M)-response curves in endothelium-denuded rat aorta. Inhibitor concentrations were selected as the concentrations previously shown to be effective in similar preparations [8,11-14]. Subsequent concentrations of dexmedetomidine were added after the previous concentration had elicited a sustained and stable contraction for 5 min. Each inhibitor was added to the organ bath 20 min before the addition of dexmedetomidine [8].
Finally, we examined the dependence of dexmedetomidine-induced contraction on extracellular calcium. Endothelium-denuded aortic rings were incubated in standard Krebs solution, low-calcium Krebs solution (1 mM CaCl2), and calcium-free Krebs solution containing 1 mM ethylene glycolbis (β-aminoethyl ether)-N,N,N',N',-tetraacetic acid for 15 min before the addition of dexmedetomidine.
Reagents
All drugs were of the highest purity available commercially. Rauwolscine, verapamil, acetylcholine, U-73122, Gd3+, L-NAME and indomethacin were obtained from Sigma-Aldrich (St Louis, MO, USA), and 2-APB was from Calbiochem (San Diego, CA, USA). Dexmedetomidine was donated by Orion Pharma (Turku, Finland). 2-APB was dissolved in dimethylsulfoxide (DMSO; final organ bath concentration: 0.1% DMSO). Unless otherwise stated, all other drugs were dissolved and diluted in distilled water.
Data analyses
Values are expressed as the mean ± SD. Contractile responses to dexmedetomidine are expressed as the percentage of the relevant maximum contraction in response to isotonic 30 mM KCl. The logarithm of the drug concentration eliciting 50% of the maximum contractile response (ED50) was calculated using nonlinear regression analysis by fitting the concentration-response relationship for dexmedetomidine to a sigmoidal curve using commercially available software (Prism version 5.00; GraphPad Software, San Diego, CA, USA). Statistical analyses for the comparison of maximal contraction and the ED50 between the no-drug group and the drug (inhibitor, low calcium, and calcium-free Krebs solution)-treated groups were performed using a one-way analysis of variance (ANOVA) by Tukey's multiple comparison test or Student's t test for paired samples. Responses to each concentration of dexmedetomidine were compared by repeated measures ANOVA followed by Bonferroni post-hoc test. P values less than 0.05 were considered significant.
Results
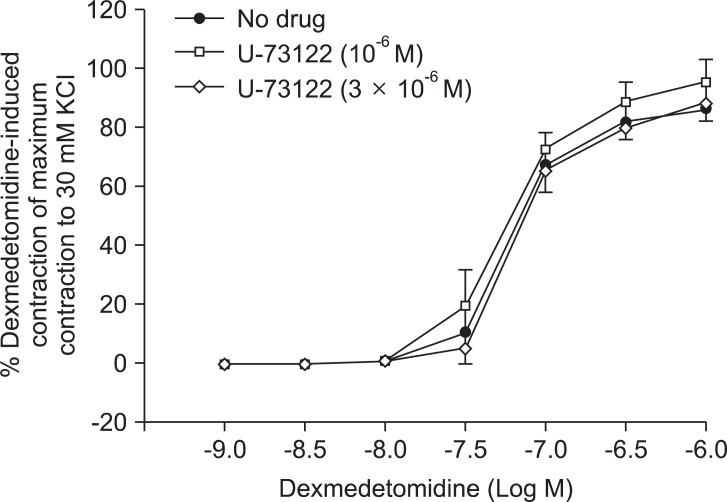
Dexmedetomidine produced concentration-dependent contraction (3 × 10-8 to 10-6 M: P < 0.001 vs. 10-9 M) (Fig. 1). Rauwolscine (10-5 M), verapamil (5 × 10-5 M), 2-APB (5 × 10-5 M), Gd3+ (5 × 10-6 M), and U-73122 (3 × 10-6 M) did not significantly alter baseline resting tension.
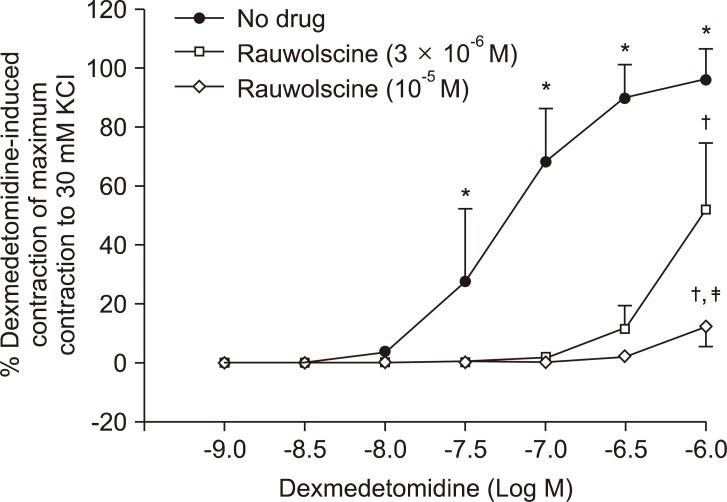
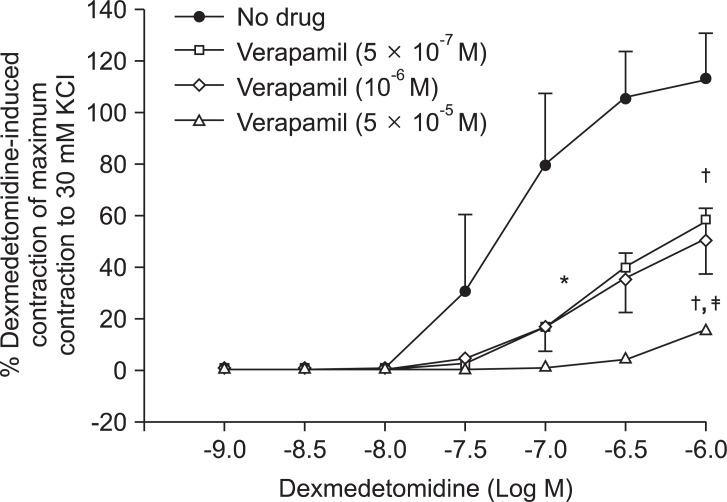
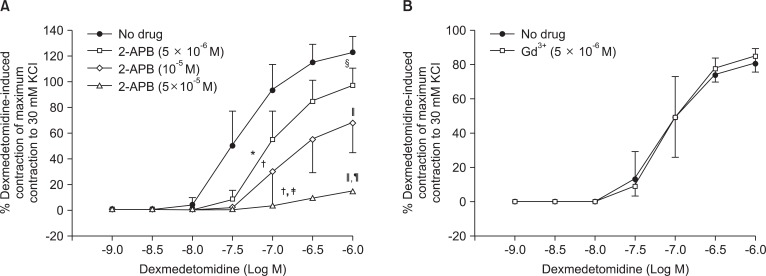
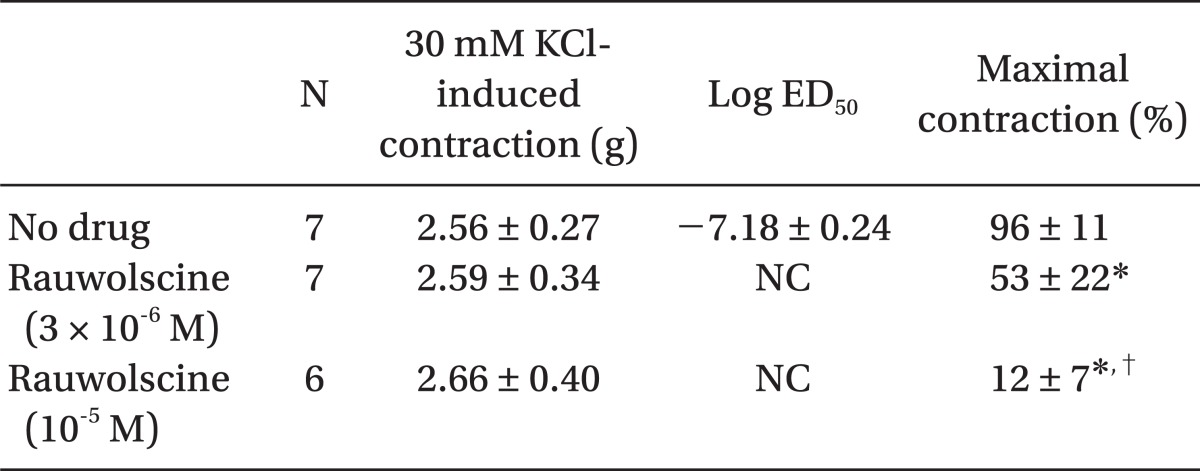
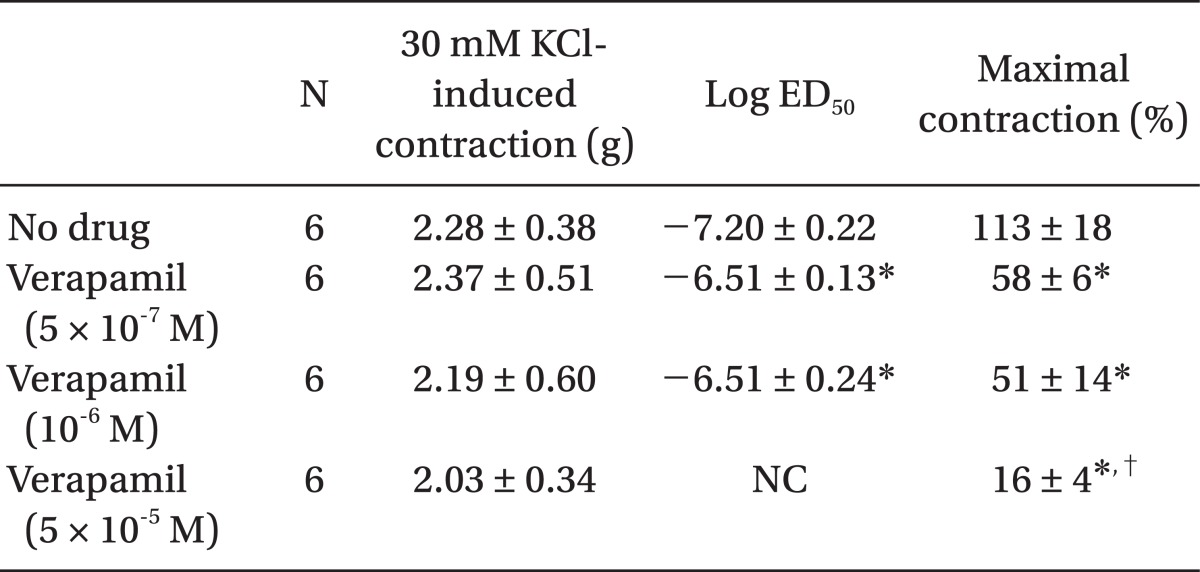
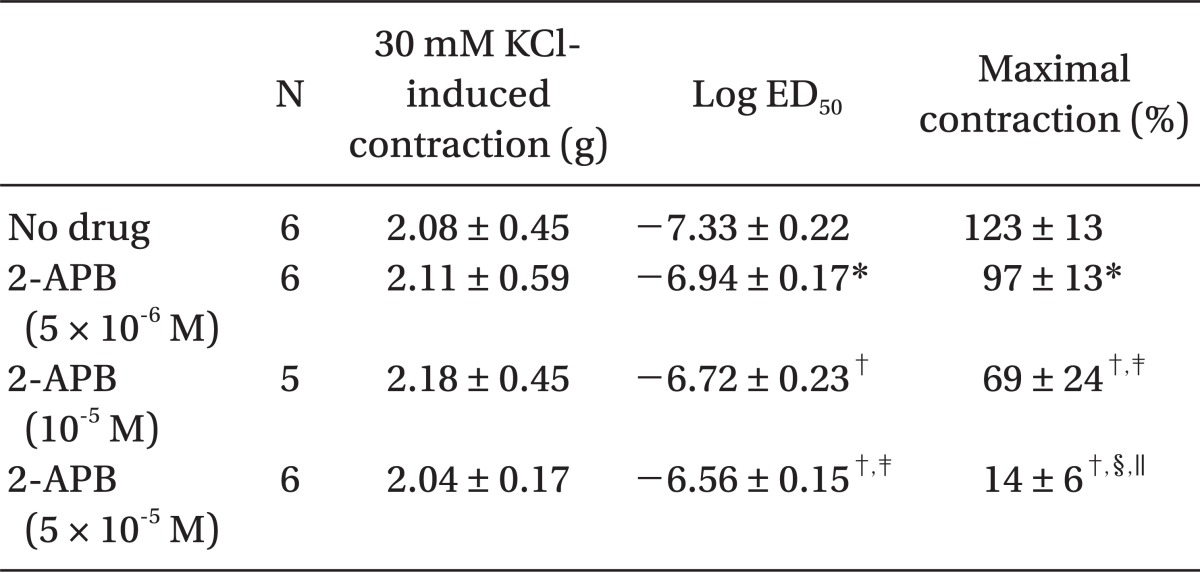
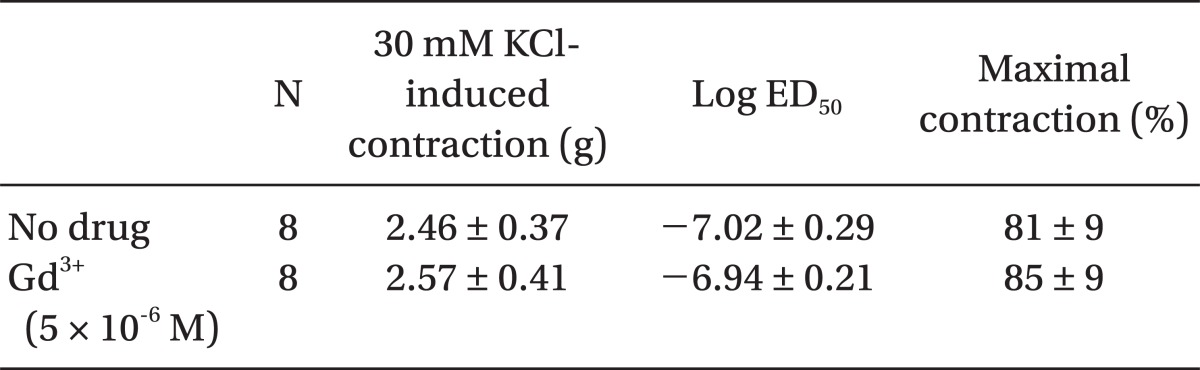
Pretreatment with rauwolscine (3 × 10-6 and 10-5 M) attenuated dexmedetomidine-induced contraction in a concentration-dependent manner (maximal concentration: P < 0.001 vs. no drug) (Fig. 1 and Table 1). Pretreatment with verapamil (5 × 10-7, 10-6, and 5 × 10-5 M) attenuated dexmedetomidine-induced contraction in a concentration-dependent manner (maximal contraction: P < 0.001 vs. no drug) (Fig. 2 and Table 2). Pretreatment with 2-APB (5 × 10-6, 10-5, and 5 × 10-5 M) attenuated dexmedetomidine-induced maximal contraction in a concentration-dependent manner (P < 0.05 vs. no drug) (Fig. 3A and Table 3), whereas pretreatment with Gd3+ (5 × 10-6 M) had no effect on dexmedetomidine-induced contraction (Fig. 3B and Table 4).
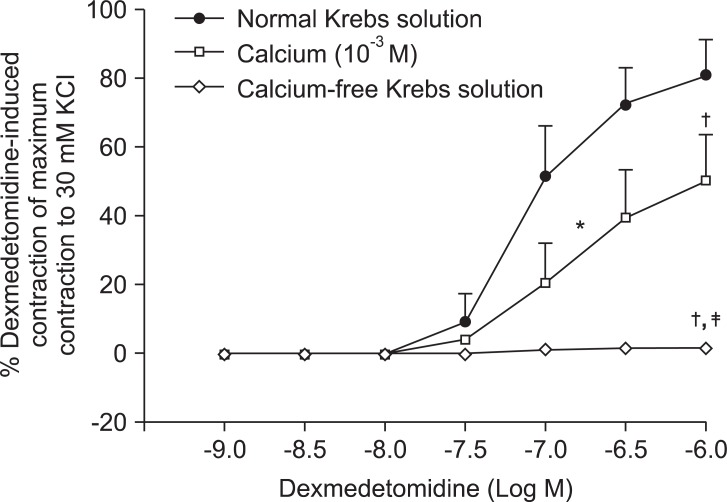
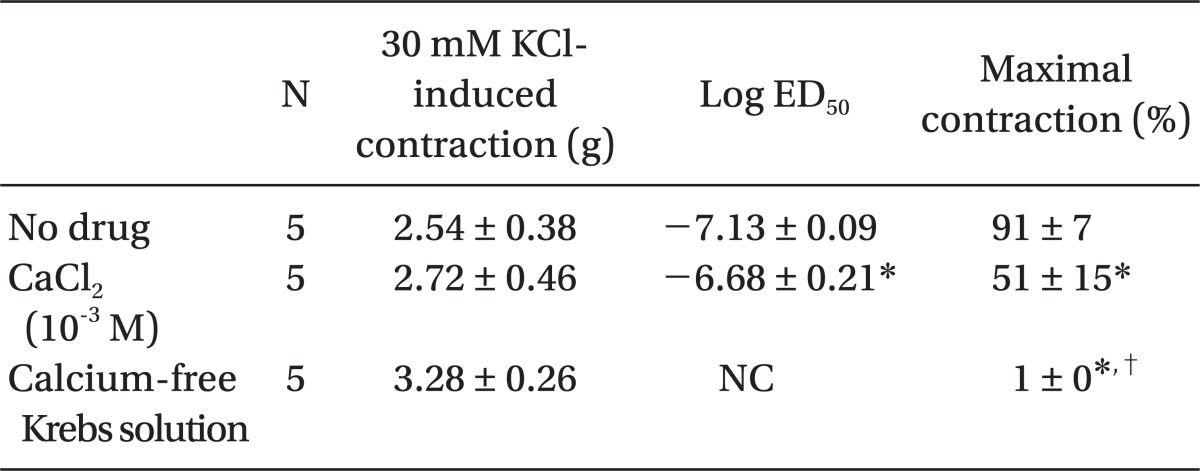
Low concentrations of extracellular calcium (1 mM CaCl2) attenuated dexmedetomidine-induced maximal contraction (P < 0.001 vs. normal Krebs solution) (Fig. 4 and Table 5). In addition, calcium-free Krebs solution nearly abolished dexmedetomidine-induced maximal contraction compared with low CaCl2 concentration (1 mM) alone (P < 0.001) (Fig 4. and Table 5).
Discussion
This study provides new information suggesting that dexmedetomidine-induced contraction is primarily dependent on calcium influx via voltage-operated calcium channels from the extracellular space in isolated endothelium-denuded rat aorta. The major findings of this in vitro study are as follows: 1) verapamil and rauwolscine attenuated dexmedetomidine-induced contraction in a concentration-dependent manner; 2) low calcium concentrations and calcium-free Krebs solution attenuated dexmedetomidine-induced contraction; 3) 2-APB attenuated dexmedetomidine-induced contraction, whereas Gd3+ had no effect on dexmedetomidine-induced contraction; and 4) U-73112 did not significantly alter dexmedetomidine-induced contraction.
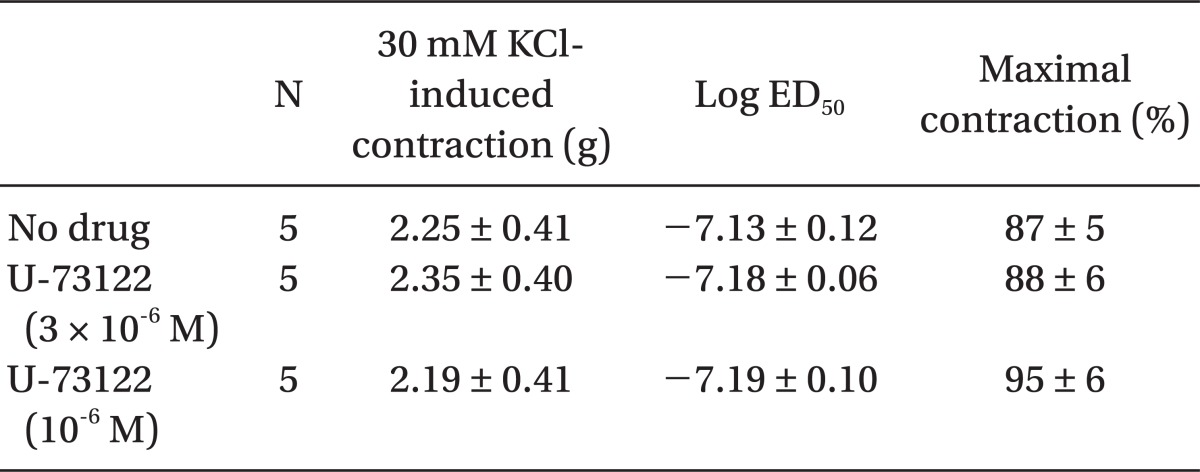
Plasmalemmal calcium channels involved in the modulation of vascular smooth contraction include voltage-operated, receptor-operated, and store-operated calcium channels [5]. At the half-maximal inhibitory concentration reported in a previous study and at concentrations exceeding the clinically relevant plasma concentration (1.32 × 10-8 M) [11,15], verapamil attenuates dexmedetomidine-induced contraction in a concentration-dependent manner, suggesting that dexmedetomidine-induced contraction involves voltage-operated calcium channel activation. Verapamil (5 × 10-5 M) severely attenuated dexmedetomidine-induced contraction. This result is similar to previous reports that voltage-operated calcium channel blockers attenuate α2-adrenoceptor-mediated contraction of isolated vessels as well as α2-adrenoceptor-mediated calcium influx of myometrial cells [8,9,16-18]. The cellular signaling pathway for α2-adrenoceptor-mediated contraction of isolated human cutaneous resistance vessel involves calcium influx via voltage-operated calcium channel activation through a pertussis toxin-sensitive G protein [17]. Phospholipase C mediates various cellular responses, including α2-adrenoceptor-mediated endothelial nitric oxide release, which is induced by dexmedetomidine [19,20]. α2A-Adrenoceptor-induced L-type calcium channel stimulation of rat portal vein myocytes involves the phosphatidylcholine-phospholipase C pathway but may not involve the phosphatidylinositol-phospholipase C pathway [21]. Similar to a previous report [21], U-73122 did not significantly alter contraction induced by dexmedetomidine with both a full α2B-adrenoceptor agonistic effect and a partial α2A-adrenoceptor agonistic effect, suggesting that dexmedetomidine-induced contraction does not involve the activation of the phospholipase C-mediated pathway. However, U-73122 inhibits α2A-adrenoceptor-mediated calcium release in porcine myometrial cells, suggesting that phospholipase C involvement of α2-adrenoceptor-mediated intracellular calcium increase may be dependent on receptor subtype affinity, species, and location [18]. Further studies regarding the signaling pathways upstream of calcium influx via voltage-operated calcium channels activated by dexmedetomidine are needed. Clonidine-induced contractile responses of isolated rat aorta are dependent on extracellular calcium concentrations [16,22,23]. In agreement with these previous reports [16,22,23], low calcium concentration (1 mM) attenuates dexmedetomidine-induced contraction, and calcium-free Krebs solution nearly abolishes dexmedetomidine-induced contraction. In addition, dexmedetomidine also increases intracellular free calcium concentrations [8], which may be associated with calcium influx from the extracellular space. Taken together, these results suggest that contractile responses induced by dexmedetomidine appear to be primarily dependent on extracellular calcium concentrations.
2-APB at concentrations that block IP3 receptor-mediated calcium release from the sarcoplasmic reticulum attenuated dexmedetomidine-induced contraction in a concentration-dependent manner, suggesting that dexmedetomidine-induced contraction may involve IP3 receptor-mediated calcium release from the sarcoplasmic reticulum [12,13]. However, 2-APB is not a highly selective blocker of the IP3 receptor, because 2-APB also inhibits store-operated calcium channels in cell membranes [24]. Thus, we investigated the effect of Gd3+ on dexmedetomidine-induced contraction because we cannot rule out that 2-APB-mediated inhibition of dexmedetomidine-induced contraction may be associated with the inhibition of store-operated calcium channels. However, the store-operated calcium channel inhibitor Gd3+ had no effect on dexmedetomidine-induced contraction, suggesting that dexmedetomidine-induced contraction does not involve calcium influx through store-operated calcium channels. Taken together, these results suggest that 2-APB-induced inhibition of contraction induced by dexmedetomidine is associated with calcium release from the sarcoplasmic reticulum. Further studies regarding the signal transduction pathway associated with dexmedetomidine-induced calcium release are needed to elucidate the detailed mechanism.
Atipamezole and rauwolscine also attenuate dexmedetomidine-induced contraction of isolated rat aorta [8-10]. In agreement with previous reports [8-10], rauwolscine attenuated dexmedetomidine-induced contraction, suggesting that dexmedetomidine-induced contraction involves α2-adrenoceptor activation.
Dexmedetomidine transiently increases blood pressure followed by hypotension in both humans and animals [3,4]. This initial, transient hypertension may be associated with dexmedetomidine-induced α2-adrenoceptor stimulation in vascular smooth muscle [6,7]. The time-to-peak serum concentration after oral administration of dexmedetomidine is delayed to a greater extent than after its intravenous administration [25]. In addition, buccal, and oral dexmedetomidine do not produce initial, transient hypertension compared with intravenous dexmedetomidine, which may be due to the delayed time-to-peak serum concentration of dexmedetomidine after its non-intravenous administration [26,27]. Considering these previous reports [25-27], the initial, transient hypertension after intravenous administration of dexmedetomidine may be due to time differences between dexmedetomidine-induced α2-adrenoceptor stimulation of vascular smooth muscle and dexmedetomidine-induced α2-adrenoceptor stimulation of the central nervous system. Clinically relevant concentrations of dexmedetomidine (10-8 M) for sedation in humans did not produce vasoconstriction in the current in vitro study, whereas supraclinical concentrations (3 × 10-8 M) of dexmedetomidine produced vasoconstriction [28]. However, any clinical implications of dexmedetomidine on regional hemodynamics must be tempered due to the fact that the aorta, regarded as a conduit vessel, was used in this study since small resistance arterioles with diameters less than 300 µm control organ blood flow. Even with this limitation, dexmedetomidine-induced vasoconstriction at supraclinical concentrations may contribute to high dose, dexmedetomidine-induced hypertension observed in previous in vivo studies [29,30]. In addition, our results suggest that the hypotension observed at therapeutic plasma concentrations of dexmedetomidine may not be mediated by the direct effects of dexmedetomidine on blood vessels but rather by an indirect vasodilatory action associated with both central and peripheral sympathetic inhibition [28].
In conclusion, dexmedetomidine-induced contraction at supraclinical concentrations (3 × 10-8 to 10-6 M) is primarily dependent on extracellular calcium concentrations and is strongly associated with calcium influx via voltage-operated calcium channels of isolated endothelium-denuded rat aorta. Dexmedetomidine-induced contraction is mediated via α2-adrenoceptor stimulation and appears to be partially mediated by calcium release from the sarcoplasmic reticulum.
 XML Download
XML Download