

 PDF
PDF Citation
Citation Print
Print


Introduction
Cytochrome c (Cyt c) is a ubiquitous, heme-containing protein that normally resides in the space between the inner and outer mitochondrial membranes. The classical function of Cyt c in the intermembrane space is as a component of the electron transport chain and leading to the reduction of oxygen to water. In 1996, another role for Cyt c as an apoptogenic agent was reported [1]. The significance of Cyt c to the apoptotic process was revealed by the findings that mitochondrial released Cyt c combines with apoptosis protease activating factor-1 (Apaf-1), procaspase-9, and dATP in the cytosol. Thereby, producing active caspase-9 [2,3]. As apoptosis has been implicated in a number of CNS disorders, including neurodegenerative diseases and damages as a consequence of stroke or CNS trauma, pharmacological prevention of Cyt c release from mitochondria is being intensely investigated as an approach with potential to reduce significantly the clinical manifestations of apoptosis in the CNS.
Neuropathic pain results from pathology in the nervous system. By modulating both synaptic inflow and dorsal horn neuronal excitability, presynaptic and postsynaptic inhibition regulates the transfer of information from the periphery to the CNS. In neuropathic pain, noxious stimulation sensitizes central neurons involved in processing of nociceptive information and this sensitization induces nocuous responsiveness to innocuous peripheral stimulation. This phenomenon is called "central sensitizatio". Reduction or elimination of spinal cord inhibition could be expected and sensitization of higher order neurons is a key mechanism involved in generating pathological excitability, that is to say the development of neuropathic pain.
Partial sciatic nerve injury (PSNI) [4] is a well-characterized mononeuropathy model to study neuropathic pain caused by nerve injury. Interest has been recently focused on the intracellular transduction pathway, involved in the transsynaptic effects of nerve injury, especially in the GABAergic inhibitory interneuron. In the present study, focuses were on some of the pieces of evidence from results that support a role for mitochondrial Cyt c release in acute neurological injury and chronic neurodegeneration. The authors investigate the effects of the PSNI-induced neuropathic pain model in mitochondrial Cyt c release and the effects of cyclosporine A (CSA) that has neuroprotection on its mitochondria stabilizing activity in PSNI.
Materials and Methods
Male Sprague-Dawley rats weighing 180-200 g were used. Rats were housed with free access to food and water under a natural day/night cycle. Rats were acclimated for 7 days before any experimental procedures. All experiments were conducted during the lights-on period in a quiet room. Animal care was provided in adherence to guidelines of the Institutional Care and Use Committee at Pusan National University Hospital.
Two sets of experiments were performed. Firstly, the animals (total n = 30) were randomly assigned to one of the 2 groups and each group received different operations (Group P; rats received PSNI operation, Group S; rats received sham operation, n = 15 per group). PSNI model was performed as described previously by Seltzer et al. [4]. Briefly, the rats were anesthetized with 3 vol% sevoflurane and 97% oxygen and placed in a prone position. An incision was made into the left thigh. After the thigh muscle was pushed aside under the dissection microscope, the sciatic nerve bundle was exposed, and then tightly ligated a half of nerve with 6-0 silk thread. The wounds were closed. Sham-operated control rats underwent an identical surgical procedure as the PSNI rats, except that the sciatic nerve bundle was without ligation.
For western blot and immunohistochemical staining, five rats in each groups were deeply anesthetized and killed at 1, 3, and 7 days after the operation. The spinal cords that included L4-5 were removed and immediately frozen on dry ice. Spinal cord tissue was homogenized in buffer (250 mM sucrose, 20 mM Tris-HCL [pH 7.4], 1.5 mM Na-EDTA, 1.5 mM Na-EGTA, 1 mM MgCl2, 1 mM DTT [Sigma-Aldrich, St. Louis, USA], 20 mM KCl [Samchun Pure Chemical, Pyeongtaek, Korea]) and centrifuged. The supernatant was decanted and used for western blot analyses. The protein concentration of the supernatant was measured using a BCA protein assay kit (Pierce, Rockford, USA). The supernatant, with equal amounts of 50 µg protein, was then fractionated by sodium dodecyl sulfate-polyacryl amide gel electrophoresis (SDS-PAGE) gel and transferred onto a nitrocellulose membrane (Millipore, Bedford, USA). Membrane were blocked with 2% BSA in Tris-buffered saline containing 0.1% Tween (TBST) at room temperature for 1 h and then incubated overnight at 4℃ with antibody against β-actin and Cyt c. After washing with TBST, anti-rabbit IgG and anti-mouse IgG2B was used as a secondary antibody (1 : 2,000 dilution in 2% BSA in TBST, 1 h 30 min incubation) and the antigen antibody complexes were visualized using an enhanced chemiluminescence detection reagent (Amersham). Bands were scanned using a densitometer (GS-700; Bio-Rad Laboratories), and quantification was performed using Multi-Analyst 1.0.2 software (blue color).
The spinal cords (L4-5) were sectioned transversely at a thickness of 6 µm on a freezing microtome for immunohistochemical staining. One tissue from each rats were then dehydrated for 2 h and fixed for 15 min in cold acetone. After washed by PBS and non-specific antibody binding was inhibited by incubation for 1 hour in blocking solution (1% BSA, 0.2% powdered skim milk, 0.3% Triton-X 100 in PBS). Primary antibodies were diluted in PBS blocking buffer and slides were incubated overnight at 4℃ in primary antibodies to GABAergic cells antibody (abCam, Share, UK). After washed by PBS, secondary antibodies Alexa Fluor 633 anti-rabbit IgG (Invitrogen, Oregon, USA; blue color) were used to locate the GABAergic cells in each section. The nuclei were visualized using Leica TCS SP2 (Leica Microsystems Heidelberg GmbH, Germany). For fluorescence intensity analysis, LCS light software (Leica Microsystems Heidelberg GmbH, Germany) was used. The staining was visualized using an optical microscope, the appearance of GABAergic cells was assessed at X1600 and fluorescence intensity was assessed at X400.
In a second set of experiments, the animals (total n = 40) were randomly assigned to one of 2 treatment groups. Depending upon group designation, the animals received CSA (Group C, n = 20) or saline (Group S, n = 20) at pre-operation, postoperative 1 day, 3 day and 7 day. CSA injected intraperitoneally in a solution of polyethylene glycol/sterile saline/cremophor oil solution (20 mg/kg). The rats of group S were treated with the 1 ml of saline in order to serve as controls.
Behavioral tests were conducted blindly by the experimenter who did not know the nature of the experimental manipulation and performed on 5 rats per group at preoperation, postoperative day 1, day 3 and day 7. The foot withdrawal threshold to mechanical stimuli (mechanical threshold) was used as an indicator of mechanical sensitivity of the affected paw. Before drugs injected to rats, behavioral tests were performed. The foot withdrawal threshold to mechanical stimuli was assessed by 8 calibrated von Frey hairs. For each test, the animal was placed in a plastic chamber (15 × 20 × 30 cm) and habituated for at least 15 min. The chamber was placed on top of a mesh screen so that mechanical stimuli could be administered to the plantar surface of left hind paw. Thresholds were determined by the up-down method [5], using a set of von Frey monofilaments. Eight von Frey filaments with bending forces of approximately equal logarithmic increments (0.22) were chosen (equivalent to 0.41, 0.70, 1.20, 2.00, 3.63, 5.50, 8.50, and 15.10 g, respectively). Starting with the 2.0 g hair, the filaments were applied perpendicular to the ventral surface of the base at the proximal part of the third or fourth toe for 6 s. An abrupt withdrawal of the foot during stimulation or immediately after stimulus removal was counted as a positive response. Whenever a positive response occurred, the next smaller von Frey filament was applied. Whenever a negative response occurred, the next higher filament was applied. The test was continued until the third change in response to stimuli had been obtained. The 50% threshold value was calculated using Dixon's formula [6].
Cyt c and GABAergic neuron were evaluated by the western blot method and immunohistochemical staining as described in first experiment (n = 15 per group).
All data are presented as means ± standard errors of the mean (SEM) and analyzed using the PASW 18K statistical program (SPSS institute, USA). Mechanical thresholds, Cyt c and fluorescence intensities of sections were analyzed with the Mann-Whitney U test to compare values between groups at each time point. In all cases, a P value < 0.05 was considered to be significant.
Results
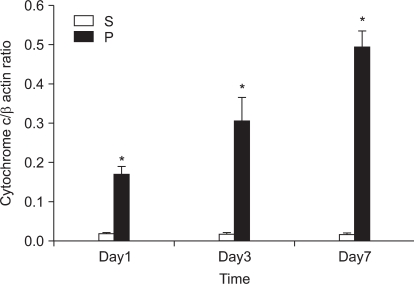
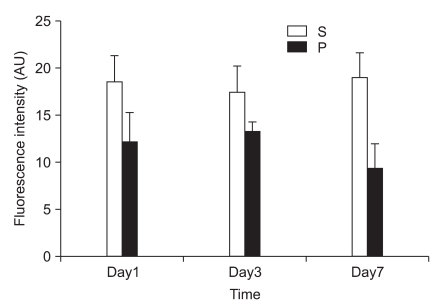
In the first experiment, postoperative foot drop was not observed. In group P, the nerve damage associated with PSNI elicited behavioral features characteristic of neuropathic pain: the rats developed abnormal gait, posture, guarding and protecting behavior and licking of the hind paw of the ipsilateral side of sciatic ligation after 1 day of operation. The rats did not put weight on the affected side and the hind leg of the affected side was drawn close to the body with distinctive guarding posture. The foot was ventroflexed and the toes were held together tightly. PSNI of the sciatic nerve in the rat produced a profound and prolonged decrease of mechanical thresholds within 1 day of the nerve injury. As shown in Fig. 1, Cyt c of group P was increased compared with group S (P = 0.020, 0.021 and 0.021) in the L4-5 at 1, 3 and 7 days after PSNI operation. The number of neurons in the ipsilateral L4-5 dorsal horn (laminae I-III) was not reduced between all groups at 1, 3, and 7 days after operation (X1600). To confirm the reliability and specificity of the neuronal marker, fluorescence intensities of these sections were examined by electron microscope (X400) and the median of group P was not significantly different from group S at 1, 3, and 7 days after operation (Fig. 2).
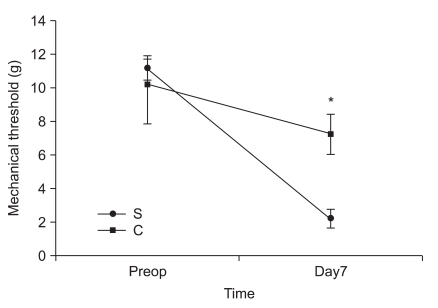
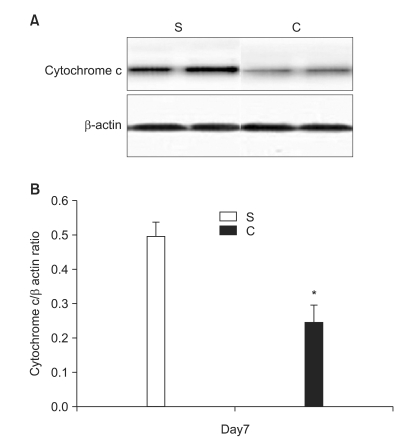
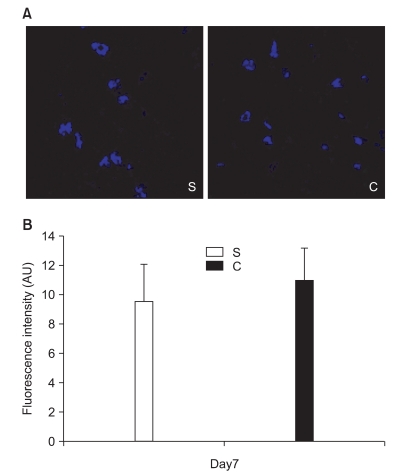
In the second experiment, the mechanical thresholds of group C were high compared with that of group S after 7 days (P = 0.009) and significant anti-allodynic effect occurred on the 7th day after PSNI surgery (Fig. 3). The Cyt c level of group S was significantly greater than that of group C at 7th day after PSNI surgery (P = 0.021). A significant reduction of Cyt c by CSA was shown after PSNI operation (Fig. 4). But same as the first experiment, there was no significant difference between group S and group C in GABAergic neuron cells and fluorescence intensities at 7 days after PSNI (Fig. 5).
Discussion
In the present study, we demonstrated that PSNI in rats increased the release of cytosolic Cyt c from mitochondria. However, GABAergic cells were not decreased in the spinal cord level on the ipsilateral side to the PSNI. The results of second experiment reveal a dramatic reduction in Cyt c release using CSA in PSNI model. Rats receiving CSA were afforded the antiallodynia without decrease of GABAergic cell.
A great advance in our understanding of the mechanisms that underlie neuropathic pain states has come from the development of animal models. Since most of these models involve partial nerve injury, some input is preserved. This allows behavioral analysis of changes in thermal and mechanical withdrawal thresholds, in addition to the assessment of spontaneous pain and autotomy. PSNI model not only produces profound behavioral indicators of persistent pain, but it also initiates a cascade of events resulting in altered peptide expression in the superficial dorsal horn [7]. Although the relationship of these changes is unclear, it has been suggested that alterations in peptide expression represent adaptive responses to promote survival and recovery of the injured neurons [8]. In the present study, there were a significant difference in the level of Cyt c between sham and PSNI model in the L4-5 segment, and moreover the effects of CSA which decreased Cyt c released from mitochondria compared with S group. These results suggest that PSNI-induced mechanical allodynia is associated more with release Cyt c from mitochondria.
Recognition that mitochondria could release triggers of cell death, and that at least one of these triggers, Cyt c, was sufficient for the latter stages of cell death, highlighted the potential gatekeeper role of mitochondria in cell death cascades [9]. The link between mitochondrial function and the transduction of cell death signals presumably involves interactions between mitochondrial free radical production and Ca2+ homeostasis [10,11]. This dysfunction appears to be dependent upon the sequestration of Ca2+ by mitochondria, given the fact that inhibition of mitochondrial Ca2+ uptake is neuroprotective. By inhibiting the opening of the mitochondrial permeability transition pore (MPTP), via its interaction with matrix cyclophilin, CSA maintains mitochondrial homeostasis. The modulation of the MPTP, a non-specific pore of the inner mitochondrial membrane, by CSA is well documented and has been shown to decrease in efficacy as a function of time [12-14]. In the central nervous system, mitochondrial involvement seems essential for normal expression of cell death phenotypes, and interference with these pathways thus seems a reasonable approach to neuroprotection. The MPTP plays an important role in cell death since it opens, leading to mitochondrial swelling and release of Cyt c, which initiates apoptosis. In first experiment, significant increase the ratio of Cyt c in P group after PNSI operation was showed and second experiment showed CSA attenuated the decrease of mechanical threshold and the increase the ratio of Cyt c. These findings suggest that the release of Cyt c from mitochondria was a result from PSNI and CSA decreased release of Cyt c of upper neuronal cell.
Although MPTP inhibitors are suggested to be protective in some models [15], inhibition of Cyt c release was not demonstrated in most cases and non-MPTP effects were not excluded, such as protective general inhibition of protein synthesis [16]. Detailed analysis using both biochemical and imaging techniques have excluded a role for the MPTP in the mechanism of Bax-induced Cyt c release from isolated brain mitochondria [11,17]. Nevertheless, the MPTP may still play a role in neuronal cell death, perhaps by signaling Bax translocation to mitochondria [18], participating in intracellular Ca2+ or ROS signaling, or regulating the release of larger mitochondrial apoptogenic proteins that occurs downstream of Cyt c release. Recently it was shown that both Bax and Bak are dispensable for NMDA-induced death of cerebellar granule neurons [19], perhaps suggesting a comparatively greater role for calcium in forms of neuronal death that are not purely apoptotic.
The use of CSA is associated with numerous side effects. Neurotoxicity was previously less well-known, but with growing experiences, central nervous system side effects are now reported. The rate of neurotoxicity appears to be dose related and increases with additional doses of chemotherapy delivered [13]. Unfortunately, neurotoxicity due to CSA can occur at normal dose and at high dose of drug. In contrast, CSA greatly induces the release of these neurotransmitters at low concentrations [20] and have neuroprotective effects.
A permanent decrease or loss of spinal inhibition after nerve injury is a potential mechanism of neuropathic pain. This mechanism that has been proposed to contribute to neuropathic pain is peripheral nerve injury-induced loss of the GABAergic inhibitory interneuronal system in the spinal cord [21]. The GABAergic inhibitory interneuronal system has an important role in the modulation of the noxious stimulation transmitted from the primary afferent input. Some studies revealed the role of GABAergic inhibitory interneuronal system in the modulation of the pain transmission and the apoptotic changes of the GABAergic interneurons in the neuropathic pain [7]. On the contrary to this, there is also evidence to suggest that death of GABAergic neurons does not occur after nerve injury. Polgar et al. [22] showed that there was no loss of neurons and no change in the proportion that were GABA-immunoreactive, in laminae I-III of the ipsilateral dorsal horn in CCI model. They also found those 4 weeks after spared nerve injury (SNI) the number of neurons in laminae I-III was not altered and that the apoptotic cells seen in the spinal cord at earlier stages were microglia, rather than neurons. However, even if GABAergic neurons do not undergo apoptosis, loss of inhibition after nerve injury could still result from depletion of GABA from their axon terminals, leading to reduction of transmitter release, and thus the size of inhibitory postsynaptic currents [23].
In conclusion, the present study suggests that mitochondrial release of Cyt c probably contributes to nerve dysfunction after PSNI. This study provides evidence that PSNI induced neuropathic pain was profoundly linked to mitochondrial stabilization, but it is presumed that pathology of PSNI is not related to the loss of neurons in dorsal horn lamina in the rats. Thus, in agreement with previous evidence, the potent neuroprotector, CSA, might produce antiallodynia through its capability to inhibit the opening of MPTP.
 XML Download
XML Download