

 PDF
PDF Citation
Citation Print
Print


Historical Views on Human Subject Research
Studies on human are imperative for medical progress and have expanded our understanding and capability to treat serious diseases and entities. However, research with humans needs to take into account the ethical dimensions of the reasons for running an experiment and the proper procedural steps to ensure that the results reflect good science. Protecting human participants in research is our top priority and has been given great consideration in the ethical conduct of research because the exact risks and benefits of research are uncertain.
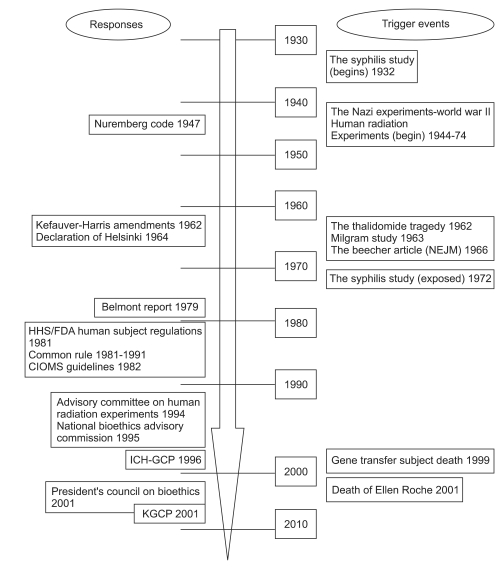
"All human beings are born free and equal in dignity and rights. They are endowed with reason and conscience and should act towards one another in a spirit of brotherhood" (Article 1 of the United Nations Universal Declaration of Human Rights). These rights have often been ignored in public perceptions of human research. Beginning in the seventeenth century, the scientific revolution brought about a method of investigation using controlled observation and reporting of result to the public as proof. The numbers of participants involved in early experiments were small and most often included the researchers themselves or their families. The most typical and famous example of this was when Edward Jenner tested a smallpox vaccine on his son and on the neighborhood children in the early modern times. The progression to the current status of protecting human research participants has been the consequence of historical events in the twentieth century. There have been many groundbreaking events that have affected the public's perception of human clinical research. The history of human subject abuses, scandals, tragedies and the responses to them are shown in Fig. 1 in chronological order.
World War II
In Imperial Japan Army Units 731, 1644, 1855, 8604 (China), 9420 (Singapore), Japanese doctors conducted live experiments with dissection, dismemberment, and bacteria inoculation on prisoners of war. They induced epidemics on a large scale, with an estimated 3,000 to 200,000 Chinese, Korean, Mongolians, and Allied civilians becoming infected [1,2]. Many prisoners were killed, directly or indirectly, by these experiments. After the war, the Supreme Commander of the Allied Powers in Japan, Douglas MacArthur, gave immunity in the name of the United States to Shiro Ishii and all members in exchange for protecting the results from the Soviet Union. No formal investigation or trial took place in association with the Japanese experiments. In the meantime, the Nazis were placing victims in vacuum chambers with low air pressure and a lack of oxygen in order to determine the health effects on pilots at extremely high altitudes. Subjects were immersed for hours in tubs of ice water, fed nothing but salt water for days, and experimented upon with techniques for battlefield medicine. At the end of the war, 23 Nazi doctors and scientists were put on trial in Nuremberg from December 9, 1946 to August 20, 1947 for the unethical treatment of concentration camp inmates, who were often used as research subjects with fatal consequences. Seven were sentenced to death. A set of standards known as the Nuremberg Code was used for evaluating and judging the defendants.
The nuremberg code and the declaration of Helsinki
The Nuremberg Code comprises such principles as informed consent and absence of coercion; properly articulated scientific experimentation; and beneficence towards experiment participants [2]. The code states that : 1) Voluntary informed consent is essential without any coercion; 2) Human experiments should be designed and based upon prior animal experimentation; 3) Expected scientific outcomes should justify the experiments; 4) The experiment should be conducted only by qualified scientists; 5) The experiment should be conducted in a way that avoids all unnecessary physical and mental suffering and injury; 6) There should be no expectation of death or disabling injury from the experiment. In 1953, the World Medical Association (WMA) was provoked to make drafts that would apply the Nuremberg Code to the practice of human experiment in the medical community. Known as the Declaration of Helsinki, it was an expansion upon the Nuremberg Code and was first adopted in 1964. It has been revised several times (1975, 1983, 1989, 1996, 2000 and most recently in 2008) according to the modern ethical theory and current clinical and research practice. A prominent point of difference from the Nuremberg Code was the flexibility of the conditions of consent, which was 'absolutely essential' under the Nuremberg code. Research was permitted without consent where proxy consent, such as that of a legal guardian, was available. The Declaration of Helsinki introduced the concept of an independent committee, which evolved into the institutional review board (IRB) system used in the US [1]. The Declaration of Helsinki focuses on a systematic approach, including IRB review, unlike the Nuremberg code, which focused on the responsibility of the individual scientist, had no legal enforcement and was applied only to non-therapeutic clinical research. The Declaration of Helsinki is an important document in the history of research ethics as the first significant effort of the medical community to regulate research itself. It forms the basis of most subsequent documents and is now widely accepted as the cornerstone document of human research ethics.
The Beecher article
Dr. Henry K. Beecher, an anesthesiologist, reported 22 studies describing violations of serious ethical principles in the New England Journal of Medicine in 1966 after the publication of the Declaration of Helsinki [1,3]. This article sparked a debate on research ethics in the US. His examples were not cited simply to blame individuals but with the hope that it would call attention to abuses, in order to correct them. The experiments that Beecher cited demonstrated ethical abuses. Here are two examples: number 7 - this study on cyclopropane anesthesia and cardiac arrhythmia involved 31 patients. Carbon dioxide was injected into the closed respiratory system until cardiac arrhythmias appeared. Toxic levels of carbon dioxide were achieved and maintained for considerable periods, causing various pathologic arrhythmias. Number 17 - live cancer cells were intradermally injected without consent into 22 chronically ill, debilitated non-cancer patients for a study of immunity to cancer (Jewish Chronic Disease Hospital Case, 1963). The physicians "did not wish to stir up any unnecessary anxieties in the patients" who had "phobia and ignorance" about cancer, so they did not tell the subjects that the injection contained cancer cells.
The Tuskegee study and the Belmont report
The Tuskegee syphilis study was an infamous clinical experiment undertaken by the U.S. Public Health Service, which would later become the Centers for Disease Control and Prevention (CDC), to study the natural progression of untreated syphilis between 1932 and 1972 in Tuskegee, Alabama. The study was designed to demonstrate the need for establishing syphilis treatment programs by investigating the effects of untreated disease. A total of 399 poor, rural black men were enrolled, under the impression that they were receiving free health care from the U.S. government. Select research participants were given free medical care, meals, and free burial insurance. However, they were never told they had syphilis, nor were they ever treated for disease. In spite of the wide use of penicillin as a curative treatment for syphilis by 1951, treatment continued to be withheld from the research subjects. The announcement of the Declaration of Helsinki in 1964 had no effect on the study. Jean Heller, an Associated Press reporter, published a story about the study in the New York Times and the Washington Star on July 25, 1972. The public reaction was great and Senator Edward Kennedy held hearings about these experiments on human subjects. The syphilis study was stopped, and treatment was given to the survivors in 1973. President Clinton officially apologized to the research subjects and their families in 1997. Congress passed a National Act in 1974 creating the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. The National Commission published the so-called "Belmont Report" in 1979, which is a landmark of ethical principles in human research. The three fundamental ethical principles for using any human subjects for research are: 1) Respect for persons: protecting the autonomy of all people and treating them with courtesy and respect; this is applied in the informed consent process. Researchers must be truthful and conduct no deception; 2) Beneficence: incorporating the philosophy of "Do no harm" while maximizing benefits for the research project and minimizing risks to the research subjects is applied through risk/benefit assessments; 3) Justice: ensuring reasonable, non-exploitative, and well-considered procedures are administered fairly and equally and applied to the selection of research subjects. These principles are comprehensive and are stated to understand the ethical issue. The three principles cannot always be applied so as to solve beyond dispute particular ethical problems, however, and provide an analytical framework that will guide the resolution of ethical problems arising from research involving human subjects. Today, the Belmont Report continues to be an essential reference for institutional review boards (IRBs) and remains the basis of human subject protection regulations.
Human radiation experiments
Eileen Welsome revealed to the Albuquerque Tribune in 1993 that researchers injected plutonium into unknown subjects to study the effects of the atomic bomb under government sponsorship [2]. In 1944, President Clinton formed the Advisory Committee on Human Radiation Experiments (ACHRE) to investigate human radiation experiments and decide upon ethical and scientific standards for evaluating these events. The Advisory Committee found that several thousand governments had sponsored human radiation experiments, intentionally releasing radiation on hundreds of occasions from 1944 to 1974. The Nuremberg Code and the Declaration of Helsinki appear to have been disregarded during Cold War radiation experiments.
Thalidomide tragedy
Thalidomide was approved in Europe as a sedative drug in the late 1950s and sold in a number of countries around the world from 1957 until 1961. It was withdrawn from the market after being found to have caused birth defects in 10,000 to 20,000 children. The FDA had not approved the drug but U.S. physicians had studied its safety and efficacy. The drugs had the side effects of shrinking blood vessels and disrupting the normal development of the vessels, affecting development of the arms and legs. It was extremely damaging to the fetus if taken in the first trimester of pregnancy. In the congressional hearing with Senator Hubert Humphrey from 1959 to 1962, it was found that many people who were taking the unapproved drugs were neither informed that they were being given an experimental substance nor asked for their consent. This led to the passage of the Drug Amendments sometimes referred to as the Kefauver-Harris Amendments. Since the episode with thalidomide, researchers have been required to inform subjects of a drug's experimental nature and to receive their consent before beginning trials.
The Milgram study
The Milgram experiment (1963) was a series of social psychology experiments conducted by Yale University psychologist Stanley Milgram after reading about the Nazi Holocaust. The study was intended to measure the willingness of participants' obedience to the authorized person who instructed them to perform acts that conflicted with their personal conscience. Volunteers were recruited for a study of "memory and learning". The volunteer was to play role of "teacher" and was required to ask the "learner" questions and administer punishment via an electric shock when the learner gave wrong answer. In reality, there were no electric shocks to the learner, but they pretended to receive an electric shock. Two-thirds of the volunteers were persuaded by the investigator to administer shocks up to the highest level of 450 volts. Upon completion of the experiment, the investigator explained the deception. The focus of Mailgram's investigation was the psychological stress induced by the experiment upon the volunteers, the deception involved and the lack of true informed consent. As a result of this controversial study, the conditions of deception in human research were limited, and now need careful IRB approval.
Hepatitis in retarded children
Experiments were designed to track the development of the viral infection of hepatitis and subsequently to test the effects of gamma globulin in preventing or ameliorating the disease from 1963 through 1966 at the Willowbrook State School, a New York State institution for mentally retarded children [4]. The subjects, all children, were purposely infected with the hepatitis virus; early subjects were fed extracts from the stool of infected individuals and later subjects received injections of more purified virus preparations. This Hospital did not admit new patients after 1964, unless their parents consented to the experiment. This case drew public condemnation because of the perception that parents and their children were given little choice about whether or not to participate in research and for performing an experiment on either a normal or a mentally retarded child when no benefit can result for the children.
San Antonio contraceptive study and Tearoom trade study
San Antonio contraceptive study: In 1971, an oral contraceptive study was conducted on 70 poor Mexican-American women to evaluate the efficacy of different kinds of female contraceptive pills. A number of indigent Hispanic women, who had no way of getting contraceptives, came to a clinic seeking contraceptives. They agreed to participate in a study to determine the side-effects of contraceptives. The randomized half received oral contraceptives and the others a placebo. The two halves were switched in the middle of the study. They were not informed that they were subjects of this kind of research or that they might receive inactive medication. As expected, there were high numbers of unplanned pregnancies in the placebo group; ten of the 76 participants became pregnant during the study.
Tearoom trade study: Anonymous male homosexual encounters in public restrooms (a practice that was known as "tea-rooming" in US gay slang) were studied in a controversial 1970 Ph.D. dissertation and book titled "Tearoom trade: a study of homosexual encounters in public places" by Laud Humphreys. Humphreys, as social scientist, acted as a watcher outside public toilets where people grouped to engage in anonymous homosexual activity. He copied down license plate numbers and other identifying information, which he used to get the names and addresses of over 100 men who had been involved in 50 sex acts (mostly oral sex). He then personally visited their homes to interview them about their milieu and family life. Many subjects were living with a family in a situation where it would be upsetting to disclose their homosexual activity. At no time were the subjects informed that they were participating in a study about male homosexuality. In his published reports, the level of detail was such that the identification of some of his subjects was revealed.
Death of Jesse Gelsinger
Jesse Gelsinger, an 18-year-old volunteer, was the first person publicly identified as having died in a clinical trial of a gene transfer experiment in 1999 [5]. He suffered from ornithine transcarbamylase deficiency, an X-linked genetic disease of the liver, the symptoms of which include an inability to metabolize ammonia - a byproduct of protein breakdown. He was injected with an adenoviral vector carrying a corrected gene to test the safety of the procedure and died four days later, despite not being sick before the experiment. The principal investigator and the University of Pennsylvania shared in a private startup company that owned the technology used in the experiment. The main issue in this research was conflict of interest (COI). Moreover, investigators did not pay attention to animal data indicating the possibility of adenovirus-induced liver failure and the possible harm to Jesse's already abnormal liver function. Investigators did not use the IRB-approved consent form and had reported instances of mild liver toxicity in previous participants as adverse events.
Death of Ellen Roche
Ellen Roche, a healthy 24-year-old volunteer in an asthma study, died in 2001 because she inhaled hexamethonium, a medication used for treating high blood pressure in the 1950s and 60s [5]. She developed a cough and her condition worsened over the next week until she was put on a ventilator with progressive multi-organ failure. She was a technician from the Johns Hopkins Asthma and Allergy Center who volunteered to participate in a study designed to provoke a mild asthma attack in order to help doctors discover the reflex that protects the lungs of healthy people against asthma attacks. She died about a month after taking part in the study. Although both a National Institute of Health (NIH) and the IRB had approved the study, hexamethonium was not approved as medication by the Food and Drug Administration (FDA). A federal investigation found serious problem with IRB reviews at the University and accused the IRB of failing to take proper precautions. The IRB did not follow federal regulations and all federally funded research was suspended. Other universities were shocked and began to strengthen their IRB committees. The public expressed outrage at this case, which was readily understandable. The culture of possibly putting coercive pressure on Asthma and Allergy Center employees to participate was pointed out as a grave mistake.
International Ethical Guidelines for Human Subjects
The Council for International Organizations of Medical Sciences (CIOMS) in Collaboration with the World Health Organization (WHO) guidelines
The CIOMS (http://www.cioms.ch/) is an international, non-government, not-for-profit organization established jointly by WHO and UNESCO in 1949 to serve the scientific interests of the general international biomedical community, and has been active in dispersing guidelines for the ethical conduct of research. The international ethics guidelines created in 1993 by CIOMS and updated in 2002 for biomedical research including human subjects were intended to guide investigators from more technically advanced countries when conducting research in developing countries. The guidelines were intended to supplement alleged omissions from the Nuremberg Code and the Declaration of Helsinki, particularly when applied to cross-cultural study. The CIOMS guidelines take into account cultural differences in ethical standards. The CIOMS 21 guidelines (15 in the original report) address issues including informed consent, standards for external review, recruitment of participants, and more. The guidelines are general instructions and principles of ethical biomedical research, and have been revised to account for the latest ideas and practices, such as the Declaration of Helsinki.
The International Conference on Harmonization-Good Clinical Practice (ICH-GCP) guidelines
The ICH (http://www.ich.org/) is composed of expert working groups from the pharmaceutical industry and regulatory authorities in the European Union, Japan and the United States, as well as those of Australia, Canada, the Nordic countries and the World Health Organization (WHO). The goal is to discuss the scientific and technical aspects of drug registration and published guidelines for GCP in response to the increasingly global face of drug development, so that the benefits of international harmonization for better global health can be realized worldwide. The objective of the ICH-GCP (Geneva: 1996) guidelines is to provide a unified standard for the European Union (EU), Japan and the United States to facilitate the mutual acceptance of clinical data by the regulatory authorities. Thus, any country that adopts this guideline technically follows this same standard. Clinical studies should be carried out according to International Conference on Harmonization (ICH)/WHO Good Clinical Practice standards. This worldwide GCP document offers standardization for clinical trials of drugs. Standards for the design, conducting, analyzing, monitoring, auditing, recording, and reporting of clinical trials provide assurance that the data and reported results are credible and accurate, and that the rights, integrity, and confidentiality of trial subjects are protected. Ethical and scientific quality standards for designing, conducting, recording and reporting trials that involve the participation of human subjects ensure that the rights, safety and well-being of the trial subjects are protected. GCPs are consistent with the ethical principles originated in the Declaration of Helsinki. The ICH topics are divided into four categories (Q: quality topics, S: safety topics, E: efficacy topics E6 (R1: Revision 1) - Good Clinical practice, M: multidisciplinary topics) and ICH topic codes are assigned according to these categories. The ICH-GCP includes the following sections: (Section 1): Glossary, (Section 2): The Principles of ICH-GCP, (Section 3): Institutional Review Board/Independent Ethics Committee (IRB/IEC), (Section 4): Investigator, (Section 5): Sponsor, (Section 6): Clinical Trial Protocol and Protocol Amendments, (Section 7): Investigator's Brochure, (Section 8): Essential Documents for the Conduct of a Clinical Trial. ICH-GCP, therefore, embraces all aspects of all clinical trials. KGCP (January 1, 2000) was completely revised to harmonize with ICH-GCP regarding standards for clinical trials of drugs in Korea; compliance with KGCP during clinical trials is inspected for all investigations.
Task and Responsibilities in Human Subject Research
Institution
The Institution has the responsibility to comply with the laws and guidelines regarding oversight of all human research activities, especially when the research involves vulnerable people [6]. It also has the responsibility of educating investigators on ethical issues, scientific truthfulness, preventing misconduct and conflicts of interest. The institutions are required to have 1) ethical (IRB) review of protocol and informed consent, 2) administrative review of proposals, contract and grants, 3) scientific peer review [6].
Ethical review: By compliance with the law and guidelines, the institution can guard the rights, safety and welfare of research participants. The IRB must review the following requirements in order to give approval to research: 1) the risks are rational and minimized in relation to the anticipated benefits to the subjects based on a risk/benefit analysis; 2) the choice of subjects is equitable; 3) informed consent is obtained from each potential subject or a legally responsible representative unless waived in harmony with the law and guidelines. This should be documented on the consent form; 4) when subjects are likely to be vulnerable to coercion or undue influence, additional safeguards are needed; 5) appropriate monitoring and observation with continuing review should be scheduled when collecting data to ensure the safety of the subjects, protect the privacy of participants and to maintain the confidentiality of data. The purpose of the IRB is to ensure that the investigator complies with the protocol and to demonstrate that the trial is necessary and that the risk-benefit ratio is acceptable by reviewing key trial documents to ensure that the subjects' rights and well-being are protected.
Administrative review: the research institution generally ensures that proposals and allied budgets are in compliance with the law and institutional policy including IRB review where suitable. If the researcher has a conflict of interest, the institution should make a decision as to whether the conflict can be managed. The research institution has usually established a Conflict of Interest (COI) committee to avoid and/or to minimize potential conflicts under the instruction of institutional policy.
Scientific peer review: scientific review should examine the soundness and worth of the hypothesis, the procedure to prove the hypothesis and the appropriateness of the methods to be used. It is unethical to expose subjects to unnecessary risks and sample size justifications must be back up based on the expected results and statistical significance. When the IRB plays the dual role of conducting the scientific review, scientifically qualified experts must be added to the IRB, or the IRB should establish a subcommittee for supporting scientific review.
ICH-GCPs provide protection for human volunteers and ensure the accuracy and reliability of data generated in the course of clinical trials. Compliance with these standards is a public pledge that the rights, safety and well-being of clinical trial participants will be protected. GCPs cover obtaining informed consent, documentation, reporting adverse events and proper record keeping.
Investigator
The welfare and safety of research subjects is ultimately the responsibility of the investigator. The researcher thus shares responsibility with the research institution and sponsors. Investigators must be properly qualified to conduct the research and studies must be suitably designed to produce valid results. Investigators are responsible for ensuring that research is conducted according to the research design as approved by the IRB [4,6]. Good and professional judgment is required throughout the research process to guarantee the protection of study subjects. Investigators must protect and respect the personal dignity and autonomy of the research volunteers by obtaining informed consent before a person agrees to participate in a study. Subjects are protected from harm by study proposals that maximize anticipated benefits and minimize possible risks. The benefits and burdens of research are reasonably distributed. Protecting subjects and achieving scientific progress are not exclusive and not conflicting. The principal investigator can delegate study-specific task and responsibility to other team members including subinvestigators, the Clinical Research Coordinator (CRC), as well as a variety of professionals, statistician, laboratory technicians and administrative staff. Studies should be conducted according to the protocol (study design) that the IRB approved. This is the duty of an investigator in amenability with the regulations. The protocol is a formally written document detailing how the research is to be conducted. The institution policies, guidelines and law state the items that must be included in the protocol and informed consent. The study procedures and inclusion/exclusion criteria are to be evaluated and checked while the protocol is in its draft form. The investigator ought to decide upon the feasibility of recruiting volunteers with/without advertising prior to approving a study. All changes to the protocol must be agreed upon by the IRB and sponsors before execution. Investigators should document and clarify deviances from the protocol. The detection of major or repeated noncompliance with the protocol can result in closing of the study or even ineligibility as an investigator.
Ethical Issues
Ethics in clinical design
Researchers and IRB members must carefully inspect and bear in mind the details of research design protocol such as randomization, blinding, and the problem of placebos as controls and assessment of risks and benefits.
The distinction between research and treatment
The ethics of research and therapy are fundamentally different. However, clinical research and therapy both provide medical care and are performed by physicians with similar interventions of treatment in the clinical setting [2]. Experimental interventions and the best proven therapy should appear equally effective. Physicians commonly conduct clinical research and medical therapy as intimately connected. The purpose of clinical medicine is to provide optimal medical care for individual patients; it is ethically governed by the principle of therapeutic beneficence and nonmaleficence. On the other hand, clinical research is not a therapeutic activity devoted to the personal care of patients. It is carried out to answer a scientific question with the aim of producing knowledge that can be generalized and applied to future patients. The clear demarcation between research and therapy becomes blurred when physician-investigators view patients as subjects in practice. Physicians and patients commonly fail to appreciate the distinction between research and therapy because of the similarity in the physician and patient relationship, especially with regard to the setting out of innovative or non-validated therapies. To be sure, the risks need to be assessed by physicians and patients and they must weigh carefully the options of standard treatment and research intervention, of course with the informed consent of the patient.
Clinical equipoise and randomized clinical trials (RCTs)
RCT is a study design that randomizes whether the participants are given treatment or placebo for the sake of eliminating prejudice. RCTs are ethical only in conditions of "clinical equipoise" being assured. Random selection of participation can yield scientifically convincing data for use in future patients. However, critics of RCTs say that individual therapy is determined not by the participants' physical needs and personal value but by the statistical requirements of the study design. Randomization to get data for future patients sacrifices benefits for the present patients. RCTs violate the physician's duty of giving the most appropriate treatment to their patients. One way of solving this problem is to obtain fully informed consents of the participants. Small losses in some patients might be ethically tolerated as long as the patients are not exposed to unnecessary risk. RCTs are ethically permissible using a standard of clinical equipoise in the context of non-life threatening therapies. Serious problems remain, however, in clinical equipoise that can easily be upset. So long as the study intervention is balanced, RCTs are acceptable.
Placebos in clinical research
RCTs are well recognized as the most desirable type of study to evaluate a new treatment, but many clinical trials are concerned about the use of placebos as controls. Placebo controls are intended to ascertain the authentic effectiveness of a treatment while eliminating various disturbing factors and to determine the actual therapeutic efficacy of a new treatment. If researchers wish to test a new treatment in the absence of a known effective treatment, the use of a placebo is usually problematic and unethical. Comparisons of new drugs to current standard medications and comparisons to placebos are different. The latter comparison conflicts with the Declaration of Helsinki, which requires that any new method be tested against the best existing prophylactic, diagnostic, and therapeutic method(s). Placebos can have their own powerful ambiguous effects. Comparing against placebos is not the same thing as testing against nothing. A lack of difference between a new drug treatment and the standard treatment does not necessarily mean that the new drug is effective. The new drug and the standard treatment could both be effective or both be ineffective. The standard treatment might be generally effective, but lose its effect in a particular situation. The FDA considers placebo controls to be the gold standard of measuring diagnostic or therapeutic efficacy because they rely on statistical significance in judging the efficacy of the new drug. It is likely that placebo studies will continue to be used. However, they should be used with caution so that patients do not face unnecessary pain or disease on account of a medical experiment in keeping with the ethical use of placebos in any experiment.
The ethics of phase I research
The main purpose of Phase I trials is to determine the highest tolerated dose of a new drug in humans, with the hope of gathering information that may help patients in the future. Human studies, especially phase I cancer trials, bring about much tension and conflict between the goals of science and those of clinical care, bringing special challenges to IRB review. Almost all Phase I studies are executed on normal human volunteers to determine the level of toxicity and pharmacologic effects of receiving higher doses of a drug on a small number of participants. However, studies that are conducted on sick patients, such as trials of cancer drugs, can be extremely controversial because the drugs are too toxic to be administered to a healthy volunteer. This category of patients is seriously ill and highly vulnerable. These individuals are designated to participate in phase I oncology trials for the good of society with no premeditated benefits and need special protection. Sometimes they are under the misconception that the trials are designed to help them [5]. Consent documents should detail the purpose of this trial and indicate that the dose will be increased until the patient gets extremely sick. Moreover, it is impossible to predict the side effects that the patient will experience because the study is designed to push the dose of the study drug until toxicity is unacceptable. Despite this, most participants think that the main purpose of trial is to make them better. Information including the purpose, risks and benefits of the study should be provided to make clear the distinction between research and patient care. Standardized wording should be required on these consent documents.
Participant recruitment
Clinical trials should be conducted with the willingness and generosity of those who serve as human participants. Recruitment is almost inevitably time-consuming, expensive, and requiring of the investigator's realistic determination of its feasibility prior to performing the trials. Many patients still have the idea that clinical trials are treatment, especially when they have serious disease. Investigators should guard against exaggerating the benefits of research and should ensure realistic assessments of the benefits and risks before volunteering their patients to become subjects. Concerns prior to participation are the fear of receiving a placebo instead of the active drug, as well as the risky side effects. The fact that research participants are supererogatory volunteers means that investigators and physicians should sustain heavy responsibilities not to violate their trust. People should be selected to make sure that the burdens and potential benefits are equitably dispersed. It is ethically justified to exclude those at greater risk of injury. Therefore, after careful selection of subjects best able to answer the scientific questions and to understand the risks and potential benefits posed by that particular trial, participants are identified, recruited and enrolled according to their eligibility criteria. The scientific and ethical basis of including women and minorities in clinical research are that many have begun to see access to clinical research and to test drugs as an advantage rather than a burden from which people should be protected. Some even saw their participation in the research as not only beneficial, but as essential to their medical care and their chance of survival. On the other hand, once recruitment and enrollment of participants with appropriate inclusion and exclusion criteria have been decided, one controversial problem is the amount of money to pay. Payment should be prohibited, although compensation for expenses may be ethically permitted.
Informed consent
The voluntary consent of the participant in a clinical trial is now an indispensible part of human research. The process need to include the three key components of information, understanding and voluntary agreement, in order to be ethically suitable. The firmest foundations for the requirement to seek consent are based upon the ethical principle of respect of persons described in the Belmont Report. These imply that individuals should be treated as self-ruling agents and that person with diminished autonomy should be protected. Participating subjects will be treated as an end and not merely as a means to another's end, based on Kantian terms. However, informing the prospective subject that a clinical trial will be at least in part a means is a consent issue in human research that differs from practice. Only emergency and therapeutic concession exceptions are allowed in the context of medical practice. In cases of emergency or life-threatening situations, informed consent can be impossible to get and can sometimes cause postponement of asking the consent of the subject or permission. There is continued controversy over deferred consent as privileges [7]. The therapeutic exception to withholding information is when disclosure would be harmful to the patient's interest or well-being. The subject might be invited to consent to incomplete disclosure with the promise of full disclosure at the termination of the research. Fully informed consent is an ideal goal that we can never achieve, but we must attempt to reach it. Competence and comprehension to reach an enlightened decision is the domain of controversy. Many studies involve unreal or uncertain benefits and the subject's participant represents only a societal good. We need to provide subjects the opportunity to choose what is best for themselves in order to gain their trust while also taking into account the ethical issues of consent.
International research
A vital issue in international research is exploitation in developing countries. In most developing countries, obtaining voluntary and informed consent is problematic, making it difficult to conduct studies [8]. Many trials that make use of impoverished populations in developing countries violate the most fundamental understanding of ethical attitudes. However, researchers insist that doing research with placebo-controlled studies in developing countries is at least equivalent to the standard of care in these countries, which consists of unverified regimens or no treatment at all. It is now ethically acceptable to most that researchers working in developing country have a responsibility to provide treatment that conforms to the standard of care in the sponsoring country, and, when possible, to resolve the double standard between developing and developed countries. Cultural relativism or community beliefs cannot be used as a justification for violating universal human rights. There must be a core list of human rights that must be protected despite local distinctions in their superficial features. Ethical standards in medicine similarly cannot be relative. The force of local customs or law cannot justify abuses of certain fundamental rights, and the right of self-determination based on informed consent. When researchers from developed countries collaborate on studies performed in developing countries, it is important to stick to these fundamental principles to avoid ethical imperialism and to justify studies. There is an enormous amount of research to be done in developing countries, with their diverse and large populations and the burden of public healthcare that has yet to be solved. A truly international effort is needed to relieve the populations that have suffered so dreadfully. A collaborative effort will be required to conduct ethically and scientifically sound research that yields solid results.
Other issues
Remaining issues include special populations, genetics research, stored human biological specimens, human embryos and stem cells, drug challenges and drug washout studies, research with communities, scientific misconduct, behavior of clinical investigators, conflicts of interest, research with secondary subjects, tissue studies and records review, and behavioral research issues [4]. These issues are not presented here due to lack of space, but need to be debated. They have not been excluded here because they are any less important than those discussed above.
Criticisms to the IRB System and Suggestions
IRB review is the main body of research supervision, making IRBs the key protectors of human research participants. However, concerns have been raised about the adequacy of IRB review. In spite of the roles and responsibilities of IRBs, the fact is that many are overloaded, understaffed and faced with a variety of skeptical criticism. Many IRBs are lacking the resources and staff to carry out the hefty task of reviewing research [9,10].
IRBs have acknowledged a number of criticisms for their performance: 1) the monitoring function of IRBs ongoing research is not fulfilled on their role for annual review, consent, adherence to protocol, and data integrity. Auditing and quality assurance programs serve an important preventive role; 2) both free standing commercial review boards (non-institutional review boards), which are financially dependent on their client, and academic IRBs, the members of which are inclined to accept the studies of their colleagues, have conflicts of interest inherent in their structure. The independence and integrity of both types of IRBs should be secured to avoid problems; 3) multi-center trials by different IRBs cause delays and inconsistencies in IRB review. Exempted or expedited review at another site might be considered to eliminate duplication of effort and to reduce workload when the same study is fully reviewed at some local IRB. The central IRB model with facilitated review process could be a reasonable way to lessen the burden on local IRBs; 4) IRBs pay out too much time reviewing and revising consent forms. Usually consent forms are written at the reading level of a college graduate, and different IRBs in multi-center trials may produce inconsistent consent forms; 5) a review of the scientific benefits of the trial is often beyond the scope of the IRB.
Accreditation of IRBs may be an effective approach to improving quality, as an indicator of superiority in human subject protection. The Association for the Accreditation of Human Research Protection Programs (AAHRPP) carries out voluntary accreditation of IRBs requiring self-assessment, site visits, and evaluation. Electronic and structured forms are also suggested to reduce paper work and expedite the review process.
Conclusions
Biomedical research has made remarkable advances over the past century; as a result, ethics in clinical research is of more concern than ever before. There was little public dispute over the ethics of biomedical research until the 1960s, when scandals appeared to erupt worldwide and were opened to the public (Fig. 1). There have been many responses to these scandals including recognition of the need for standards and guidelines in the ethics of clinical research. The growing necessity for ethics in clinical research has raised concerns related to controversial issues in the processing of the formal mechanism known as the IRB. There exist various perspectives in special topics with or without consensus. This paper first introduces historically evoked scandals and responses, and then identifies key ethical issues and insights, with topics limited by space constraint. Selected debates are intended as a guide to the ethical issues confronted by physicians and researchers. Research ethics is an essential part of good research practice to protect participants in clinical studies. It is our optimistic belief that these challenging issues will be resolved through a consensus in the future. It is also my hope that this review provides an idea of the ethical framework to those investigators and anesthesiologists who will need to meet the challenges of changing patterns of research circumstances.
 XML Download
XML Download