

 PDF
PDF Citation
Citation Print
Print


Introduction
Morphine is commonly used for chest pain in acute myocardial infarction. Morphine is a nonspecific opioid receptor (OR) agonist, meaning it activates all of the OR subtypes, i.e. µ-, δ- and κ-OR subtypes. It has been proposed that morphine-induced preconditioning (M-Pre) confers cardioprotection against ischemia/reperfusion (I/R) injury and this effect is as effective as ischemic preconditioning (I-Pre) [1]. The cardioprotective effect by M-Pre involves the activation of δ-OR, especially δ1-OR [2,3]. Recently, it has been demonstrated that morphine-induced postconditioning (M-Post) is also cardioprotective in I/R-injured hearts [4]. Therefore, it is highly possible that M-Post may involve δ-OR.
Meanwhile, the mitochondrial permeability transition pore (MPTP) opening is considered an end target for the cardioprotection against I/R injury [5,6]. It has been generally accepted that the inhibition of the MPTP opening is a crucial event for cardioprotection in both I-Pre [7] and ischemic postconditioning (I-Post) [8]. Recently, it was reported that morphine prevents the MPTP opening in rat cardiomyocytes [9]. In the present study, we investigated the involvement of the δ-OR subtype and MPTP in M-Post in isolated rat hearts.
Materials and Methods
The experimental procedures and protocols used in this study were reviewed and approved by the Institutional Animal Care and Use committee of our hospital. Male Sprague-Dawley rats (270-320 g) were obtained from KOATECH Co, Cheongwon-gun, Republic of Korea. Rats were anesthetized with 100 mg/kg of pentobarbital sodium (Entobar®, Hanlim Pharmacy, Yongin, Korea), and then anticoagulated with 300 IU of heparin administered intraperitoneally. Coronary perfusion using the Langendorff system with modified Krebs-Henseleit (KH) solution, containing 118.5 NaCl, 4.7 KCl, 1.2 MgSO4, 1.8 CaCl2, 24.8 NaHCO3, 1.2 KH2PO4, and 10 glucose (in mM) was performed as described previously [10]. All hearts were allowed to stabilize for at least 20 min before induction of ischemia. Hearts were subjected to 30 min of regional ischemia and 2 h of reperfusion. To induce regional ischemia, a snare was made at the level of the proximal length of left coronary artery. Regional ischemia was induced by pulling the snare and confirmed by regional cyanosis in addition to a substantial decrease in left ventricular developed pressure (LVDP). Reperfusion was started by releasing the snare. Hearts experiencing ventricular fibrillation (VF) after reperfusion usually revert spontaneously to a sinus rhythm. VF lasting more than 30 s was treated with a finger flick cardioversion until a perfusing rhythm was obtained. No pharmacological agents were used for defibrillation. In isolated hearts, a KH buffer-filled latex balloon was inserted into the left ventricle (LV) and was adjusted to 5-10 mmHg of left ventricular end-diastolic pressure (LVEDP) at the beginning of the experiment. Cardiodynamic variables, including heart rate (HR), left ventricular systolic pressure (LVSP) and LVEDP were recorded with the BIOPAC system (BIOPAC Systems Inc., CA, USA). LVDP and rate-pressure product (RPP) were calculated as follows; LVDP = LVSP - LVEDP and RPP = LVDP × HR. Maximum of the first derivative of LV (+dP/dtmax) was analyzed using an analysis software (BSL v3.7.3.). Coronary flow (CF) was measured by collecting perfusate dripping from the right heart into a graduated cylinder.
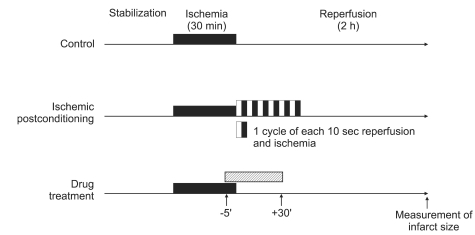
Isolated rat hearts were randomly allocated to control (CON, no other intervention either before or after index ischemia) and treated groups (Fig. 1). In the M-Post group, morphine was added to the perfusate 5 min prior to reperfusion to 30 min after reperfusion at a concentration of 1 µM. As a comparison, another group of hearts was induced with I-Post by 6 cycles each of 10 s reperfusion and 10 s global ischemia. To assess the involvement of δ-OR in M-Post, 100 µM of the non-selective OR antagonist naloxone (M-Post + NAL), 5 µM of the selective δ-OR antagonist naltrindole (M-Post + NTD), and 100 nM of the selective δ1-OR antagonist 7-benzylidenenaltrexone (M-Post + BNTX) were perfused along with morphine. The concentrations of morphine, naloxone, naltrindole and 7-benzylidenenaltrexone were chosen based on previous reports used in isolated working rat hearts [10,11]. An additional M-Post group was treated with 20, 40, or 60 µM of a MPTP opener atractyloside (M-Post+ATR) to assess the involvement of MPTP in morphine-induced cardioprotection.
Morphine, naloxone, naltrindole, and 7-benzylidenenaltrexone were purchased from Tocris Cookson Inc., Ellisville, MO, USA. Atractyloside was purchased from Sigma-Aldrich Chemical., St. Louis, MO, USA. Chemicals were dissolved with distilled water and stored at -20℃ and were diluted with KH solution to the required final concentrations on the day of each experiment.
At the end of each experiment, ischemic (area at risk, AR) and infarcted (area of necrosis, AN) regions were measured with fluorescent polymer microspheres (Duke Scientific Corp., Palo Alto, CA, USA) and 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich Chemical., St. Louis, MO, USA) staining as described previously [12]. The AR and AN regions were quantified with the program ImageTool (UTHSCSA Image Tool, version 3.0). Infarct size was expressed as a percentage of the risk zone (AN/AR). All measurements were performed in a blinded fashion. Body weight (mean 301.6 ± 1.8 g) and heart weight (mean 1.48 ± 0.01 g) were equivalent among the groups.
Data are presented as means ± SEM. Data analysis was performed with a personal computer statistical software package (SPSS for Windows, Release 12.0; SPSS Inc, Chicago, IL, USA). Data were analyzed using one-way analysis of variance (ANOVA) with a Bonferroni post-hoc test. Differences were considered to be statistically significant when P values were less than 0.05.
Results
A total of 82 rat hearts were used for infarct size measurement. Five hearts were excluded for the following reasons: a CF > 18 ml/min (n = 1), LVDP < 80 mmHg (n = 2), and sustained arrhythmia (n = 2) during stabilization. Therefore we report the data from 77 successfully completed infarct experiments.
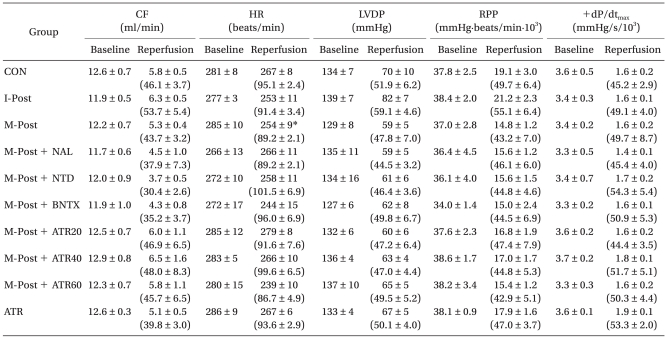
Baseline CF, HR, LVDP, RPP, and +dP/dtmax averaged 12.3 ml/min, 279.0 beats/min, 133.6 mmHg, 37.3 mmHg/min/103, and 3.5 mmHg/s/103, respectively. There were no significant differences in baseline cardiodynamic variables among the groups (Table 1). After 2 h of reperfusion, there were also no significant differences in CF, LVDP, RPP, and +dP/dtmax among the groups. In the M-Post group, however, HR was significantly decreased after 2 h of reperfusion compared to baseline levels (P < 0.01).
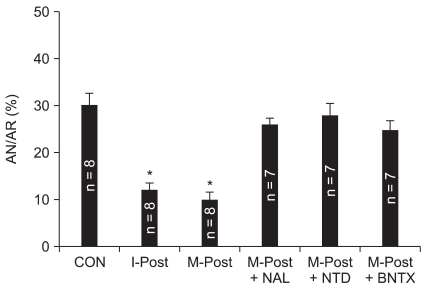
As shown in Fig. 2, infarct size in control hearts was 30.0 ± 3.7% of the AR, and this result is in agreement with our recently reported infarct measurement study [12]. Treatment with 1 µM morphine significantly reduced myocardial infarction size (9.8 ± 2.5%, P < 0.001 vs. CON). This anti-infarct effect by morphine was similar with the infarct limitation effect by I-Post (11.9 ± 2.2%, P > 0.05 vs. M-Post). Naloxone totally abrogated the infarct-limiting effect of morphine (25.7 ± 1.9%, P < 0.01 vs. M-Post). In addition, the anti-infarct effect by morphine was also totally blocked by naltrindole (27.8 ± 4.3%, P < 0.05 vs. M-Post) and 7-benzylidenenaltrexone (24.7 ± 3.7%, P < 0.01 vs. M-Post). Naloxone, naltrindole and 7-benzylidenenaltrexone themselves had no effect on infarct size (P > 0.05 vs. CON).
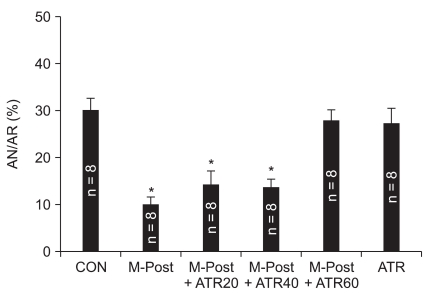
We further tested the involvement of MPTP in the morphine-induced reduction of infarct during reperfusion by using incremental doses of the MPTP opener atractyloside. Concentrations of 20 and 40 µM atractyloside could not attenuate the anti-infarct effect by morphine (Fig. 3). However, 60 µM of atractyloside (28.0 ± 3.6%, P < 0.05 vs. M-Post) totally blocked the infarct-limiting effect by morphine at a dose that had no effect in non-treated hearts (26.3 ± 5.2%, P < 0.05 vs. M-Post).
Discussion
In the present study, morphine targeting reperfusion effectively reduced myocardial infarction in isolated rat hearts and this effect mimicked the anti-infarct effect of I-Post. The infarct-reducing effect by morphine was totally inhibited by the δ-OR antagonist naltrindole and the δ1-OR antagonist 7-benzylidenenaltrexone, which suggests morphine activates the δ-OR, especially δ1-OR. In addition, the infarct-limiting effect by morphine was also totally blocked by 60 µM of the MPTP opener atractyloside, implying the involvement of MPTP in the morphine-induced anti-infarct effect. Taken together, our data strongly suggest that M-Post limits myocardial infarction via activation of δ1-OR and inhibition of MPTP opening in isolated rat hearts. After 2 h of reperfusion, there were no significant differences in cardiodynamic variables including LVDP, RPP and +dP/dtmax among the groups. However, HR was significantly decreased in the M-Post group. This may be caused by morphine's direct effect on the myocardium, causing a decrease in HR. Interestingly, the decrease in HR by M-Post group was reversed by OR antagonists and atractyloside.
Schultz et al. [1] first reported that the use of morphine to target ischemia mimicked the effect of I-Pre in open-chest rats. More recently, Gross and co-workers [4] demonstrated that the use of morphine to target reperfusion reduced the infarct size in I/R-injured rat hearts and this result is in agreement with our present study. In addition, the anti-infarct effect was as effective as I-Post in our study. Taken together, the data reinforce that morphine confers cardioprotection when administered not only during ischemia but also during reperfusion.
In adult ventricular cardiomyocytes, only δ-OR and κ-OR subtypes were reported, although more recent data suggest that there are functional µ-OR in rat cardiomyocytes [13]. It has been proposed that δ-OR is responsible for the cardioprotective effect of I-Pre and M-Pre. In our previous report, cardioprotection by I-Post involved δ-OR, especially δ1-OR in isolated rat hearts [10]. In addition, Gross et al. [14] recently reported that M-Post involves activation of δ-OR in in vivo rat hearts. Therefore, we hypothesized the possibility of involvement of δ1-OR in M-Post. In the present study, the anti-infarct effect by M-Post was totally blocked by the nonspecific δ-OR antagonist naltrindole and the δ1-OR antagonist 7-benzylidenenaltrexone. This result strongly suggests that M-Post activates δ-OR, especially δ1-OR, even though we did not evaluate the involvement of κ-OR in this study.
Chen et al. [15] reported that M-Post confers cardioprotection via activation of κ-OR but not by δ-OR. In their study, the κ-OR antagonist nor-binaltorphimine partly reversed the protective effects of M-Post, while naltrindole had no effect on M-Post. In their study, however, OR antagonists, such as naloxone, naltrindole, and nor-binaltorphimine, themselves also reduced infarct size. In addition, they induced M-Post at the onset of reperfusion while our M-Post was induced 5 min prior to reperfusion. Therefore, their results did not provide a clear distinction as to whether the infarct-limiting effect by M-Post was caused by M-Post itself or by OR antagonists. In the present study, however, our concentrations of naloxone, naltrindole and 7-benzylidenenaltrexone themselves had no effect on infarct size which suggests that the infarct-limiting effect by M-Post was caused entirely by M-Post itself.
Inhibition of MPTP is considered an end point target against myocardial I/R injury. MPTP remains closed under normal physiologic conditions but opens at the onset of reperfusion triggered by both Ca2+ overload and the excessive formation of reactive oxygen species [16]. The MPTP opening has been shown to result in both necrotic and apoptotic cell death. Inhibition of the MPTP opening in the setting of I/R injury is cardioprotective. Recently, Obame et al. [17] demonstrated that morphine-induced cardioprotection inhibited the MPTP opening by using the MPTP opener cyclosporine A as assessed by the calcein loading Co2+-quenching technique. More recently, Xi et al. [9] showed by confocal microscopy that morphine treatment prevented the MPTP opening in cardiomyocytes. We also previously reported that I-Post was mediated by inhibition of the MPTP opening via σ-opioid receptor activation [10]. The anti-infarct effect by M-Post was totally attenuated by an MPTP opener atractyloside in our isolated rat hearts, which suggests the involvement of MPTP in cardioprotection by M-Post. We used a 20 µM concentration of atractyloside based on a previous report that the anti-infarct effect by bradykinin was blocked by this concentration of atractyloside [18]. However, 20 µM and 40 µM of atractyloside could not block the infarct-limiting effect by M-Post in our present study. The infarct-reducing effect by M-Post could be blocked by 60 µM of atractyloside and this concentration of atractyloside itself had no effect on infarct size. Therefore, this result may suggest a stronger activity for M-Post to inhibit the MPTP opening than that for other cardioprotective strategies, and that higher concentrations of atractyloside may be required to block M-Post.
In conclusion, M-Post significantly reduced infarct size as compared to I-Post. The anti-infarct effect by M-Post involves δ-OR, especially δ1-OR activation and MPTP inhibition. However, the involvement of other OR subtypes, such as δ2-OR and κ-OR, and intermediate signaling pathways by M-Post should be determined in the future.
 XML Download
XML Download