

 PDF
PDF Citation
Citation Print
Print


Introduction
Since protease activation is one of known mechanisms for developing ischemic injury, inhibiting this protease activation has been proposed to attenuate post-ischemic injury [1-4]. Proteases, which are secreted from activated neutrophils during acute inflammatory phase, degrade the extracellular matrix and lead to tissue injury. While several protease inhibitors showed a myocardial protective effect in ischemia/reperfusion (I/R) injury models [5-7], ulinastatin, a protease inhibitor derived from human urine, also showed organ protective effect against I/R injury in the brain [8,9], liver [10,11], kidney [12], lung [13], and heart [6,7]. Previous investigations have showed that ulinastatin reduces the release of elastase from neutrophils and inhibits the accumulation of neutrophils in various I/R injury models [5,8,10,13] meaning that ulinastatin's protective effect is due to its anti-inflammatory response.
In this study, the effect of ulinastatin on several physiological and biochemical variables in an in vivo rat heart model of regional I/R injury was performed to determine whether ulinastatin's myocardial protective effects were due to an anti-inflammatory response.
Materials and Methods
Animal preparation
Male Sprague-Dawley rats (Sam Tako Inc., Osan, South Korea) weighing between 220 and 250 g were used for this study. All animals were maintained in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health in 1996. The protocol was approved by the Animal Research Committee.
Animal preparation and surgery were performed as previously described [14]. Animals received general anesthesia with an intramuscular injection of 15 mg/kg of tiletamine/zolazepam (Zoletil50®, Virbac Lab., Carros, France) and 8 mg/kg of xylazine (Rompun®, Bayer, Seoul, South Korea). Additionally, 15 mg/kg of tiletamine/zolazepam and 8 mg/kg of xylazine was injected to maintain the level of anesthesia after 3 h reperfusion. If the tail moved (i.e. a sign of awakening), an additional 7.5 mg/kg of tiletamine/zolazepam and 4 mg/kg of xylazine was injected to maintain the level of anesthesia. The trachea was intubated, and then lungs were mechanically ventilated (tidal volume, 8 ml/kg; respiratory rate, 80 strokes/min) with ambient air using a volume-controlled rodent ventilator (Harvard Rodent Ventilator Model 607, Harvard, Holliston, MA, USA). The arterial blood pH and gases were maintained within normal physiological limits (pH, 7.35-7.45; PaCO2, 30-40 mmHg; PaO2, 60-100 mmHg).
A paramedian sternotomy was performed, and the pericardium was opened to expose the heart. For the I/R experiments, a 4-0 black silk was passed around the left coronary artery (LCA) territory to induce regional myocardial ischemia. The coronary artery was occluded by pulling a snare through a small vinyl chloride tube and clamping it with a mosquito haemostat. Reperfusion was achieved by releasing the clamp. Myocardial ischemia was confirmed by the appearance of regional epicardial cyanosis over the myocardial surface and by arrhythmia. After 30 min of ischemia, the myocardium was reperfused by loosening the snare, which was maintained for 6 h of reperfusion. Successful reperfusion was confirmed by the disappearance of epicardial cyanosis and the production of epicardial hyperaemia and arrhythmia after releasing the snare. Body temperature was monitored with a rectal thermometer (Sirecust 1260, Siemens Medical Electronics, MA, USA) and maintained at 36-38℃ with an electrical heating pad.
Experimental protocols
The animals were divided randomly into two sets of experiments. In the first set of experiments, animals were divided into three groups as follows: (1) I/R + Normal Saline (NS) group - rats in which 1 ml NS was administered intraperitoneally 30 min before the ischemic insult (n = 10), (2) I/R + U50 group - rats in which 50,000 U/kg ulinastatin (Ulistin®, Hanlim, Seoul, South Korea) diluted with NS was administered intraperitoneally 30 min before the ischemic insult (n = 10), and (3) I/R + U100 group - rats in which 100,000 U/kg ulinastatin diluted with NS was administered intraperitoneally 30 min before the ischemic insult (n = 10). Rats underwent a 30 min ischemia followed by reperfusion of the LCA territory. Hemodynamic measurements were recorded serially (30 min, 1 h, 1 h 30 min, 2 h, 6 h) during the 6 h of reperfusion, and myocardial infarct size was measured.
In the second set of experiments, animals were divided into five groups as follows: (1) sham + NS group - rats in which 1 ml NS was administered intraperitoneally 30 min before the sham operation (n = 6), (2) sham + U100 group - rats in which 100,000 U/kg ulinastatin diluted with NS was administered intraperitoneally 30 min before the sham operation (n = 6), (3) I/R + NS group - rats in which 1 ml NS was administered intraperitoneally 30 min before the ischemic insult (n = 6), (4) I/R + U50 group - rats in which 50,000 U/kg ulinastatin diluted with NS was administered intraperitoneally 30 min before the ischemic insult (n = 6), and (5) I/R + U100 group - rats in which 100,000 U/kg ulinastatin diluted by NS was administered intraperitoneally 30 min before the ischemic insult (n = 6). All animals were exposed to 30 min ischemia followed by 6 h of reperfusion. The sham group had paramedian sternotomy and pericardiotomy performed without the LCA occlusion. The thoracic cavity was opened to expose the heart for sampling of 3 ml blood from the right ventricle. Blood samples were collected in heparin containing tubes or conical tubes to measure cardiac troponin-I (cTnI), creatine kinase (CK) concentration, tumor necrosis factor (TNF)-α, and myeloperoxidase (MPO) activity. The entire sample was centrifuged, and serum and/or plasma were collected and frozen at -70℃ until analysed.
Measurement of hemodynamic variables and myocardial contractility
The right common carotid artery was cannulated with a 2-Fr Millar catheter (Model SPR-407, Millar Instruments Inc., Houston, TX, USA) and advanced into the left ventricle (LV) to measure baseline hemodynamic function (heart rate [HR], left ventricular end diastolic pressure [LVEDP], the maximum rate of intraventricular pressure increase [+dP/dtmax], and the maximum rate of intraventricular pressure decrease [-dP/dtmax]). Hemodynamic measurements were recorded serially (30 min, 1 h, 1 h 30 min, 2 h, 6 h) during the 6 h of reperfusion.
Area at risk and infarct size determination
After cardiac function analysis, the LCA was occluded again and 2 ml of 1% Evans blue dye was injected into the pulmonary artery to facilitate area at risk (AAR) determination as described in a previous report [14]. The perfused myocardium was stained with Evans blue dye, whereas the occluded vascular bed was not. The atrium, right ventricle, and major vessels were removed from the heart, and the AAR was separated from the nonischemic area. The AAR was cut into small pieces and incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC) at 37℃ for 20 min. The non-infarcted myocardium stained a deep red color with TTC, and the infarcted area (IA) remained pale yellow. The sections were fixed for 24 h in 2% formalin, and the LV, AAR, and IA weights were determined. The AAR and IA were expressed as a percentage of the LV and AAR weights, respectively.
cTnI and CK measurements
Serum levels of cTnI were determined by an enzyme linked immunosorbent assay (ELISA) kit from Life Diagnostics (cat. no. 2010-2-HS, Life Diagnostics Inc., Wester Chester, PA, USA) as described previously [14]. The cTnI activity was detected by spectrophotometry (Infinite® F200, Tecan Group Ltd., Mannedorf, Switzerland) at 460 nm. Serum levels of CK were determined by EnzyChrom™ creatine kinase assay kit (ECPK-100, BioAssay Systems, CA, USA). CK activities were detected by colorimetric determination at 340 nm.
Measurement of MPO and TNF-α activities
Plasma level of MPO were determined by rat MPO ELISA test kit (Hbt HK 105, Hycult biotechnology b.v., Uden, Netherlands) according to the manufacturer as described previously [15]. MPO activities were detected to determine neutrophil accumulation by spectrophotometry (Infinite® F200, Tecan Group Ltd., Mannedorf, Switzerland) at 460 nm after reaction of the plasma sample at MPO antibody-coated plate. Plasma concentrations of immunoreactive TNF-α were determined with an ELISA kit (RAT00, R&D Systems inc., Minneapolis, MN, USA) according to the manufacturer's protocol as described previously [15]. This assay employs the quantitative sandwich enzyme immunoassay technique. Plasma was reacted with the assay reagents in the TNF-α kit and analyzed by spectrophotometry (Infinite® F200, Tecan Group Ltd., Männedorf, Switzerland) at an absorbance of 460 nm.
Statistical analysis
All values are expressed as the means ± SD. All statistical analyses were performed using SPSS software (ver. 12.0 for Windows, SPSS Inc., Chicago, IL, USA). The differences between groups were evaluated using a Friedman one-way analysis of variance and a Tukey's multiple comparison post hoc test. Statistical significance was defined as P < 0.05.
Results
Hemodynamic function following I/R injury
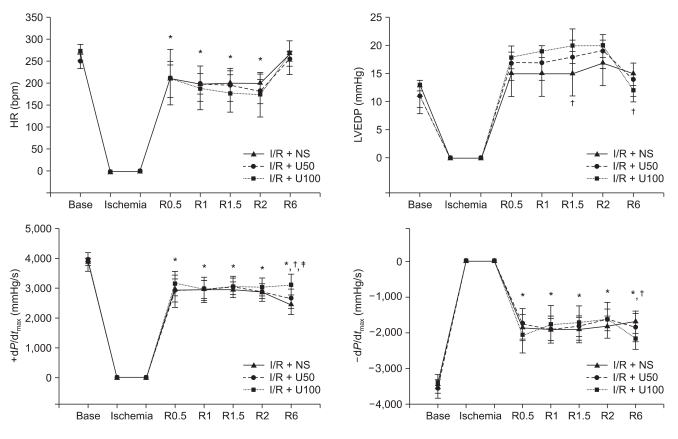
The hemodynamic measurements recorded serially during the 6 h after reperfusion revealed substantial LV dysfunction as demonstrated by decreased ±dP/dtmax after I/R injury in the I/R + NS group compared to the basal hemodynamic function (Fig. 1). The LVEDP in the I/R + U100 group (12 ± 2 mmHg, P = 0.012) showed significant difference compared to the I/R + NS group (15 ± 2 mmHg ) after 6 h of reperfusion. The ±dP/dtmax in the I/R + U100 group (+dP/dtmax: +3,088 ± 362 mmHg; P < 0.001, -dP/dtmax: -2,157 ± 290 mmHg; P = 0.011) showed significant difference compared to the I/R + NS group (+dP/dtmax: +3,972 ± 227 mmHg/sec, -dP/dtmax: -3,412 ± 248 mmHg) after 6 h of reperfusion. The HR was not significant difference between the groups.
Myocardial infarct size
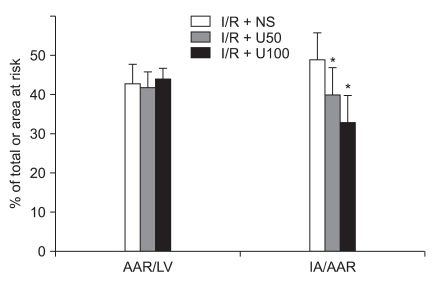
The AAR/LV induced by occluding the LCA territory was not significantlt different between the three groups. The IA/AAR in the I/R + NS group (49 ± 7 %) was significantly different compared to the I/R + U50 (40 ± 7 %, P = 0.016) and I/R + U100 groups (33 ± 7 %, P < 0.001) (Fig. 2).
Serum CK and cTnI levels
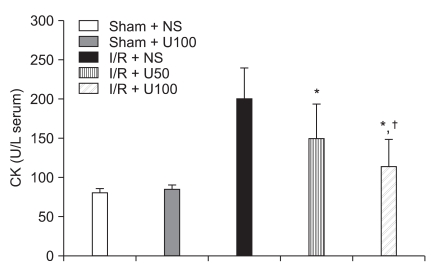
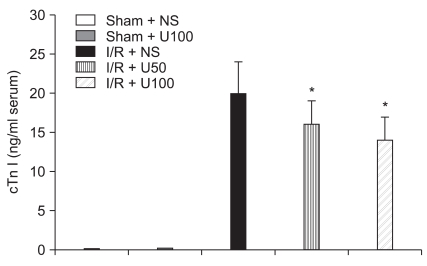
I/R induced a significant increase in the serum level of CK after 6 h reperfusion. The elevation of serum CK concentration in the I/R + NS group (200 ± 40 U/L) was significantlt inhibited in the I/R + U50 (149 ± 45 U/L, P = 0.012) and I/R + U100 (114 ± 35 U/L, P < 0.001) groups after regional I/R injury (Fig. 3). I/R induced a significant increase in serum levels of cTnI after 6 h reperfusion. The elevation of serum cTnI concentration in the I/R + NS group (20 ± 4 ng/ml) were significantly higher than in the I/R + U50 (16 ± 3 ng/ml, P = 0.043) and I/R + U100 (14 ± 4 ng/ml, P = 0.012) groups after regional I/R injury (Fig. 4).
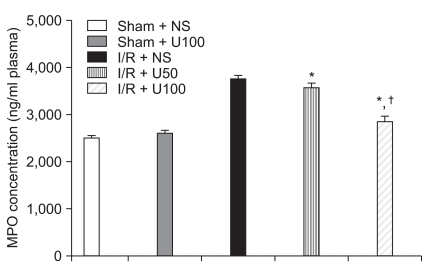
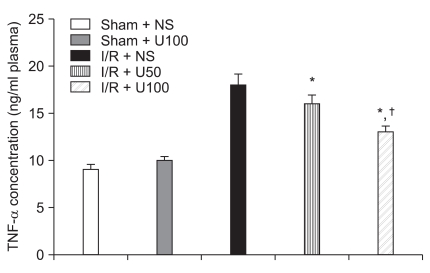
Plasma MPO and TNF-α activity
I/R induced a significant increase in the plasma level of MPO after 6 h reperfusion. The elevation of plasma MPO concentration in the I/R + NS group (3,760 ± 60 ng/ml) were significantly higher than the I/R + U50 (3,572 ± 84 ng/ml, P = 0.041) and I/R + U100 (2,848 ± 120 ng/ml, P < 0.001) groups after regional I/R injury (Fig. 5). I/R induced a significant increase in the plasma level of TNF-α after 6 h reperfusion. The elevation of plasma TNF-α in the I/R + NS group (18 ± 1.2 ng/ml) was significantly greater than the I/R + U50 (16 ± 0.9 ng/ml, P = 0.042) and I/R + U100 (13 ± 0.6 ng/ml, P < 0.001) groups after regional I/R injury (Fig. 6).
Discussion
Myocardial I/R injury is a major event in many clinical situations including the perioperative period, coronary thrombolysis, percutaneous coronary intervention, coronary artery bypass graft, and cardiac transplantation [16-18]. The non-cardiac surgical patient at risk of myocardial I/R injury is growing because of perioperative morbidity and mortality [16,17]. In this situation, applying the appropriate agents to reduce myocardial I/R injury is critical. Thus, considerable research has been made to determine means to protect against myocardial injury. This study describes the benefit of ulinastatin as a myocardial protective agent.
Ulinastatin improved myocardial contractility, reduced myocardial infarct size, and decreased serum levels of CK and cTnI after regional I/R injury in an in vivo rat heart model. This protection appears to be, at least in part, due to inhibition of TNF-α and MPO activities, which are indexes of tissue neutrophil infiltration and activation.
Myocardial infarction causes neutrophil infiltration into the infarct zone and increases the production of pro-inflammatory cytokines such as TNF-α [19-21]. Accumulation of neutrophils is evidence of acute inflammation and the infiltration of neutrophil can initiates myocardial damage [19,20]. The MPO activity assay is widely used to a biomarker for neutrophil infiltration and inflammatory reactivity, as MPO is an enzyme that is released mainly from neutrophils. Considering the attenuated MPO activity in the present study, ulinastatin may exert a myocardial protective effect by inhibiting the inflammatory process.
A previous study showed that ulinastatin has anti-inflammatory and organ protective effects in various I/R injury models [4,7-13]. Especially, ulinastatin has a myocardial protective effect by various mechanisms in an in vivo rat heart model. In reperfusion after hemorrhagic shock, ulinastatin contributed to the recovery of cardiac function after reperfusion by reducing the severity of mitochondrial dysfunction and maintaining energy production [6]. Ulinastatin also improved functional recovery of the heart after long cardioplegic arrest in an in vitro rabbit heart model [7]. In this study, ulinastatin had a myocardial protective effect via an anti-inflammatory response. Our study also showed that ulinastatin had a myocardial protective effect in the regional I/R injury in an in vivo rat heart model.
Measurements of endogenous mediator, TNF-α, allowed to elucidate the inflammatory response pathway. TNF-α is a potent pro-inflammatory cytokine that acts on vascular endothelial cells to induce expression of leukocyte adhesion molecules [20,22]. TNF-α is upregulated in the myocardium in response to a variety of cardiac injuries, including transient myocardial ischemia and reperfusion [22]. This suggests the possibility that TNF-α plays an important role in vivo in the pathogenesis of post-ischemic myocardial inflammation and I/R injury. In the present study, the significant reduction of TNF-α after ulinastatin suggests ulinastatin's possible myocardial protective effect via the inhibition of pro-inflammatory cytokine release.
Taken together, our results suggest that an anti-inflammatory effect is an important mechanism for myocardial protection following regional I/R injury in an in vivo rat heart ischemia model. The dose of ulinastatin was higher compared with clinically relevant doses. In this experiment, we used 50,000 and 100,000 U/kg of ulinastatinn because these dosages had an organ protective effect in previous studies [6,9,10].
Our study has some limitations. First, necrotic cell death reaches a peak at 24 h after I/R injury, whereas apoptotic cell death develops progressively until 72 h [23]. This study observed the myocardial protective effects after 6 h of reperfusion. Therefore, long-term observations will be needed to determine whether the effect is temporary or sustained. Second, ulinastatin was administered intraperitoneally and lacked a documented serum drug level. Therefore, this study cannot confirm that a clinically relevant ulinastatin concentration was associated with a myocardial protective effect.
In conclusion, ulinastatin reduced cardiac dysfunction, infarct size, MPO activity, and TNF-α release after regional I/R injury in an in vivo rat heart model. Its protective effect may be attributable to its anti-inflammatory.
 XML Download
XML Download