

 PDF
PDF Citation
Citation Print
Print


Introduction
It is well-established that ischemic preconditioning (I-Pre) and ischemic postconditioning (I-Post) are explicitly cardioprotective strategies. The signaling pathways of I-Pre and I-Post are becoming increasingly clear. I-Pre, a series of brief ischemia-reperfusion (I/R) cycles before index ischemia, confers resistance against a subsequent prolonged period of myocardial ischemia. The cardioprotective mechanisms of I-Pre involve autacoid factors [1], protein kinase C (PKC) [2], reactive oxygen species [3], reperfusion injury salvage kinases [4], glycogen synthase kinase-3β (GSK-3β) [5] and mitochondrial permeability transition pore (MPTP) [6], among others. I-Post, which involves brief episodes of I/R during early reperfusion, is also a powerful strategy to limit reperfusion injury. The cardioprotection by I-Post may share common pathways with I-Pre including recruitment of signal transduction pathways [7]. However, there might be limitations to clinical use of I-Pre and I-Post. Indeed, I-Pre depends crucially on intervening before the ischemic event, and it is difficult to predict the onset of acute myocardial infarction. In addition, debates on the precise I-Post algorithm to use in clinical settings are ongoing. In this regard, pharmacological strategies are possible alternative methods and may provide a feasible means of effectively producing cardioprotection clinically. During the last 2 decades, pharmacological preconditioning and postconditioning to reduce myocardial I/R injury have been extensively studied. A number of cardioprotective ligands, including erythropoietin [8], adenosine [9], bradykinin [10], statins [11] and opioids [12], have been identified. Among them opioids are particularly interesting ligands to anesthesiologists because they are widely used during general anesthesia including cardiac anesthesia. In this review, we will discuss the cardioprotective outcomes in animal experiments and human studies that used opioids, particularly morphine and remifentanil which are currently commonly used for cardiac anesthesia.
Go to : 
Evidence of involvement of opioid receptors (OPRs) in I-Pre and I-Post
After the concept of I-Pre was first described in 1986 by Murry et al. [13], a number of investigators have demonstrated that OPRs are involved in the mechanism of I-Pre-induced cardioprotection. Schultz et al. [14] first reported that OPRs play an important role in I-Pre in rat hearts. The infarct reducing effect by I-Pre elicited by three cycles of 5-min occlusion and reperfusion before 30 min ischemia was totally blocked by the nonselective OPR antagonist naloxone. One year later, Chien and Van Winkle [15] also demonstrated that the infarct limitation effect by I-Pre was blocked by (-)naloxone hydrochloride intravenously administered 25 min before index ischemia in open-chest rabbits. In addition, Schultz et al. [16] proposed that the cardioprotective effect by I-Pre was mediated by peripheral OPRs. In open-chest rat hearts, a high dose of naloxone methiodide which does not cross the blood-brain barrier antagonized the infarct sparing effect by I-Pre in their study. Taken together, these results strongly suggest that I-Pre-induced cardioprotection is likely to be OPR-mediated.
Meanwhile, there is evidence that I-Post-induced cardioprotection also involves the activation of OPRs. Zatta et al. [17] demonstrated that I-Post with 3 cycles of 10 sec reperfusion and 10 sec reocclusion at reperfusion onset increases endogenous cardiac opioids and activates local OPRs in anesthetized rat hearts. In addition, Jang et al. [18] reported that myocardial infarct size could be reduced by I-Post induced by six cycles of 10 sec reperfusion and 10 sec occlusion started immediately after reperfusion in both open and isolated rat hearts. They additionally reported that the infarct-reducing effect by I-Post was totally blocked by naloxone and the δ-OPR antagonist naltrindole. These results highly suggest that endogenous opioid peptides and OPRs are involved in the cardioprotective phenomenon of I-Post.
Go to :
Opioid peptides and OPRs
It has been well known that opioid peptide synthesis, storage and release can take place in myocardial cells. Endogenous opioid peptides are synthesized and released after myocardial stres such as hypoxia or ischemia. The three distinct families of endogenous opioid peptides, enkephalins, dynorphins, and endorphins, are derived from three separate prohormones, proenkephalin, prodynorphin, and proopiomelanocortin, respectively. The enkephalins are considered important endogenous opioid peptides responsible for cardioprotection. It has been proposed that the increase in levels of enkephalins in infarcted ventricular tissue may counteract catecholamines released during ischemia and may be a defense mechanism to minimize the size of myocardial infarction [19].
The OPRs have been shown to mediate and regulate cardiovascular system function. OPRs are present centrally (hypothalamus and brainstem) and peripherally (cardiac myocytes, nerve terminals and adrenal medulla). The OPR family comprises three primary subtypes, µ-, δ-, and κ-OPRs. Among them, δ- and κ-OPRs are present on adult ventricular myocytes of the rat [20], even though µ- and κ-OPRs are present in neonatal rat hearts [12]. The δ-OPRs have the highest affinity for enkephalins whereas κ-OPRs bind preferentially to dynorphins. These OPRs belong to the family of guanine nucleotide binding protein (G-protein)-coupled receptors (GPCRs) which are cell surface proteins that convert extracellular stimuli into cellular signals.
The δ- and κ-OPR subtypes include δ1-, δ2-OPRs and κ1-, κ2-OPRs, respectively. Multiple lines of evidence reveal that δ-OPRs, especially δ1-OPRs, play an important role in cardioprotection [16,21]. Schultz and colleagues [16] suggested that the cardioprotective effect by I-Pre was mediated via δ-OPRs because pre-administered naltrindole completely abolished the cardioprotective effect induced by I-Pre. Furthermore, this group also showed that δ1-OPRs play an important role in the cardioprotection of I-Pre because the selective δ1-OPRs antagonist 7-benzylidenenaltrexone (BNTX) but not the selective δ2-OPRs antagonist naltriben given before I-Pre significantly attenuated the infarct size by I-Pre [21]. I-Post also involves δ-OPR activation because there is evidence that the infarct-reducing effect by I-Post was totally blocked by naltrindole [18], as mentioned above.
Meanwhile, the cardioprotective role of κ-OPRs is less well characterized than that of δ-OPR and controversy continues regarding its role in myocardial I/R injury [21,22]. Schultz et al. [21] tested if the κ-OPR antagonist nor-binaltorphimine (nor-BNI) administered before I-Pre could block the antiinfarct effect of I-Pre in open-chest rat hearts. The high dose of BNTX significantly attenuated the cardioprotective effect of I-Pre while nor-BNI did not. They demonstrated that δ1-OPRs but not κ-OPRs play an important role in the cardioprotective effect of I-Pre in the rat heart. Wang et al. [23] proposed that κ-OPRs mediats the ameliorating effects of I-Pre on infarct and arrhythmia, whereas δ-OPRs mediates the effects only on infarct because nor-BNI but not naltrindole reduced both infarction and arrhythmia in I-Pre with two cycles of 5 min regional ischemia followed by 5 min reperfusion. Taken together, one can not completely rule out a role of κ-OPRs in cardioprotection.
Meanwhile, the role of µ-OPRs against myocardial I/R remains unclear because results of radioligand binding studies have disputed the presence of µ-OPRs in the myocardium. However, many studies that used morphine, which has a preference for µ-OPRs, showed a successful reperfusion mimetic. Actually, the lower dose of morphine seems to activate µ-OPRs but not δ- or κ-OPRs. Weihrauch et al. [24] demonstrated that morphine at reperfusion could reduce myocardial infarct size in rabbit hearts. Zatta et al. [17] suggested that the cardioprotective effect of I-Post appeared to involve endogenously activated µ-OPRs because the infarct sparing-effect by I-Post was abrogated by the potent µ-OPR antagonist CTAP administered at reperfusion in rat hearts. Therefore, it is plausible that µ-OPRs also may be involved in the cardioprotective mechanism.
Go to :
Exogenous activation of OPRs targeting ischemia
Over the past decade, accumulating evidence demonstrates that exogenous activation of OPRs confers cardioprotection. Exogenous administration of the naturally occurring opioid peptide Met5-enkephalin via osmotic minipump for 24 h limits myocardial infarction in anesthetized open-chest rabbits [25]. Schultz's group [26] first reported the cardioprotection by exogenously administered opioid. In their study, the exogenous pre-ischemic administration of morphine in three 5-min infusions interspersed with 5 min morphine-free periods before prolonged coronary occlusion significantly limited infarct size after myocardial I/R in open-chest rat hearts. Fryer et al. [27] demonstrated that exogenous application of the selective δ1-OPRs agonist TAN-67 15 min before index ischemia significantly reduced infarct size in open-chest rats. In addition, there is evidence that exogenous activation of δ- and κ-OPRs affords protection against myocardial stunning in isolated murine hearts. The δ-OPRs agonist BW373U86 and the κ-OPRs agonist U50,488H (trans-(±)-3-dichloro-N-methy-N-[2-(1-pyrrolidinyl) cyclohexyl]benzeneacetamide hydrochloride) administered for 10 min prior to global normothermic ischemia markedly improved post-ischemic contractile recovery as measured by left ventricular developed pressure (LVDP) [28]. Meanwhile, previous studies have demonstrated that exogenous activation of κ-OPRs with bremazocin increased myocardial infarction and another κ-OPRs agonist U50,488H exacerbated I/R arrhythmias following coronary occlusion in isolated rat hearts [29]. However, Peart et al. [22] reported that the two different κ-OPRs agonists U50,488H and ICI204,448 administered 10 min before the onset of ischemia significantly reduced infarct size in a similar extent as δ-OPRs activation by BW373U86. In addition, the κ-OPRs agonists also reduced arrhythmogenesis in their study. Recently, Maslov et al. [30] reported that peripheral δ2-OPRs activation by deltorphin II induced infarct size reduction in open-chest rat heart which suggests role for δ2-OPRs in cardioprotection. Taken together, these data verify that exogenous δ- and κ-ORs activation during ischemia indeed limited myocardial infarction and may improve functional recovery.
Go to :
Exogenous activation of OPRs targeting reperfusion
Recently, much research in myocardial I/R injury has shifted the paradigm from ischemic protection to reperfusion-mediated protection because pretreatment is seldom possible in acute myocardial infarction. It has been proposed that exogenous activation of δ-OPRs or κ-OPRs during the early reperfusion period confers cardioprotection to a similar extent as targeting the ischemic period. Exogenously applied BW373U86 administered 5 min before reperfusion mimicked the effect of I-Post or morphine-induced postconditioning in isolated rat hearts [18]. In addition, the δ-OPRs agonist [D-Ala2, D-Leu5] enkephalin (DADLE) started 5 min before reperfusion also reduced infarct size in rabbit hearts [31]. Meanwhile, Peart et al. [32] demonstrated that κ-OPR activation by U50,488H prior to reperfusion affords cardioprotection in both rat and mouse hearts. The infarct-reducing effect by U50,448H administered immediately prior to reperfusion was abolished by nor-BNI, given 10 min prior to reperfusion. U50,488H perfused 5 min before reperfusion significantly limited myocardial infarction and its mechanism was mediated by extracellular signal regulated kinase1/2 (ERK1/2) signaling pathway [33].
Go to :
Morphine-induced cardioprotection in animals
Over the past decade, accumulating data demonstrate that pharmacological preconditioning and postconditioning with commonly used opioids, such as morphine and remifentanil, confer cardioprotection in animals. Morphine is frequently used for pain treatment during acute coronary syndromes. As mentioned above, Schultz's group [26] first reported that morphine-induced preconditioning (M-Pre) mimics I-Pre. Exogenously administered morphine before index ischemia could limit infarct size in rat hearts mediated via δ-OPRs because naltrindole completely abolished the cardioprotective effect induced by morphine [34,35]. M-Pre enhances isoflurane-induced preconditioning in open-chest rat hearts [36]. The downstream signaling pathways of M-Pre involve KATP channels [26,37-39] and PKC [40].
Meanwhile, morphine-induced postconditioning (M-Post) also has a powerful cardioprotective effect, which seems to be similar to M-Pre or I-Post. Gross and co-workers [41] reported that M-Post limited infarct size in I/R-injured rat hearts. This group also demonstrated that the use of morphine and the opioid agonist methadone to manage acute and chronic pain could produce an infarct sparing effect in open-chest rat hearts [42]. Recently, Jang et al. [18] reported that the infarct sparing effect by M-Post was totally blocked by naltrindole in isolated rat hearts, implying the involvement of δ-OPRs in M-Post. More recently, Kim et al. [43] demonstrated that the involvement δ1-ORs in M-Post because the infarct limitation effect by M-Post was totally aborted by BNTX in isolated rat hearts (Fig. 1). Interestingly, however, Chen et al. [44] reported that infarct reducing effects by M-Post in isolated perfused rat hearts was significantly blocked by nor-BNI but not naltrindole, suggesting involvement of κ-OPRs but not δ-OPRs in M-Post. In their study, however, OPR antagonists, such as naloxone, naltrindole, and nor-BNI, also limited infarct size. Therefore, their results did not provide a clear distinction as to whether the infarct limitation effect by M-Post was caused by M-Post itself or by OPR antagonists. The proposed signaling mechanisms of M-Post involve nitric oxide [19], KATP channels [44,45], phosphatidylinositol-3-kinase (PI3-kinase) [24], GSK-β [41,46], mitochondrial-sensitive potassium channels [12], and MPTP [43]. For more detail on signaling pathways involved in opioid-induced cardioprotection see the reference by Schultz and Gross [47].
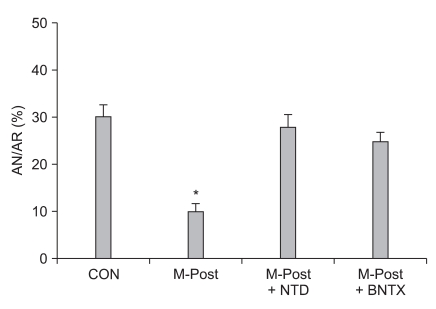 | Fig. 1Evidence for involvement of δ1-opioid receptors in morphine-induced postconditioning (M-Post). Isolated rat hearts were subjected to 30 min regional ischemia and 120 min reperfusion. Morphine (1 µM) was perfused from 5 min before reperfusion to 30 min after reperfusion. CON: untreated control hearts, NTD: δ-opioid receptors antagonist naltrindole, BNTX: δ1-opioid receptors antagonist 7-benzylidenenaltrexone. Infarct-limitation effect by M-Post was totally blocked by NTD and BNTX. *P < 0.05 vs. CON (Modified from Kim et al. [43] with permission).
|
Go to :
Remifentanil-induced cardioprotection in animals
Remifentanil, the 4-anilidopiperidine derivative of fentanyl, also is a widely used opioid along with morphine during general anesthesia. Zhang et al. [48] first reported that remifentanil-induced preconditioning (R-Pre) reduced myocardial infarct size in open-chest rat hearts. The infarct limiting effect by R-Pre was mediated via δ- and κ-OPRs. This group also demonstrated that PKC and mitochondrial KATP (mKATP) channels play an important role in R-Pre [49]. The cardioprotective mechanisms of R-Pre involve PKC, ERK1/2, and Akt pathways [50-52]. In addition, R-Pre produced delayed cardioprotection in anesthetized rat hearts and this effect was mediated via µ-, δ-, and κ-OPRs [53]. Furthermore, remifentanil-induced postconditioning (R-Post) also protects I/R induced hearts to a similar extent as I-Post which was mediated by both δ- and κ-OPRs [54] while ERK1/2 is involved in R-Post [55]. Meanwhile, remifentanil is a potent ultra short-acting synthetic opioid analgesic drug. Due to its rapid action, extremely short half-life, and rare cumulative effects, remifentanil is usually administered as a continuous infusion clinically. Therefore, it is worthwhile to determine whether remifentanil is protective only during preconditioning and postconditioning or remains protective when given continuously. However, controversy exists about the cardioprotective effect on continuous administration of remifentanil. Kuzume et al. [56] demonstrated that sustained administration of remifentanil before and throughout coronary occlusion did not result in protection of ischemic myocardium in intact rabbit hearts. However, Chun et al. [57] recently reported that continuous infusion of remifentanil targeting both ischemia and reperfusion could reduce infarct size in isolated rat hearts. The reports of pharmacological preconditioning and postconditioning with morphine and remifentanil in an isolated perfused heart, in vivo, in addition to those using cell culture models, are presented in Table 1.
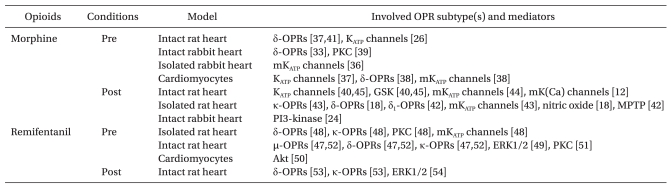
Table 1
Cardioprotection by Exogenously Applied Morphine and Remifentanil in Animal Studies
Pre: preconditioning, Post: postconditioning, OPR: opioid receptor, mKATP channels: mitochondrial KATP channels, PKC: protein kinase C, PI3-kinase: phosphatidylinositol-3-OH kinase, mK(Ca) channels: mitochondrial calcium-sensitive potassium channels, MPTP: mitochondrial permeability transition pore, ERK1/2: extracellular signal-regulated kinases, GSK: glycogen synthase kinase.
![]()
Go to :
Functional recovery by morphine and remifetanil against myocardial I/R injury
Usually, opioids have a negative chronotropic effect, i.e. decreasing heart rate (HR). M-Pre decreases HR after reperfusion in open-chest rat hearts compared to control hearts in open-chest rat hearts [58]. M-Post also decreases HR but has no significant difference in LVDP compared to control hearts [43]. R-Pre results in a decrease in HR, mean arterial blood pressure (MBP), and rate-pressure products (RPP) in open-chest rat hearts [48,52] and R-Post and continuous infusion of remifentanil during myocardial I/R also significantly reduces HR [55]. Recently, Chun et al. [57] compared five different remifentanil-based strategies, including R-Pre, R-Post, ischemic targeting remifentanil, reperfusion targeting remifentanil, or both ischemic and reperfusion targeting remifentanil, against myocardial I/R injury. Interestingly, R-Pre better preserved LVDP, which is regarded as a contractility marker of the isolated rat heart, than various other remifentanil-based treatment strategies (Fig. 2). They demonstrated that the reason for that effect might be due to the pharmacology of remifentanil and/or OPR responses. Namely, continuous infusion of remifentanil targeting only ischemia or only reperfusion may result in OPR desensitization and/or downregulation of OPR sensitization.
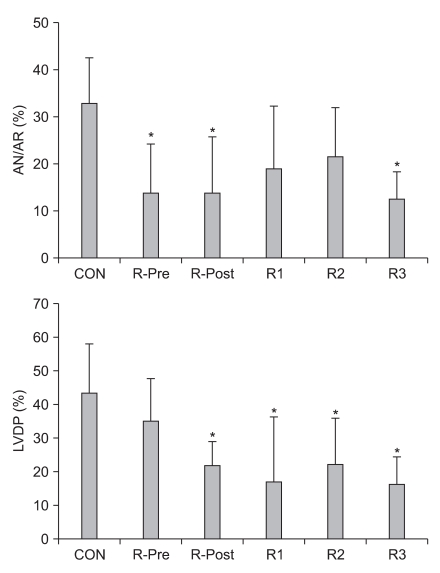 | Fig. 2(Top) Area at necrosis (AN) relative to percentage of area at risk (AR) as evaluated by triphenyltetrazolium chloride staining. (Bottom) Percentage recovery of left ventricular developed pressure (LVDP) after reperfusion in isolated rat hearts. CON: untreated control hearts, R-Pre: remifentanil preconditioning, R-Post: remifentanil postconditioning, R1: remifentanil targeting ischemia, R2: remifentanil targeting reperfusion, R3: remifentanil continuous infusion throughout ischemia and reperfusion. *P < 0.05 vs. CON (Modified from Chun et al. [57] with permission).
|
Hemodynamic changes after reperfusion by opioids are variously reported according to animal models or ischemic severity. Even in the same experimental animal model with almost identical protocols, the hemodynamic results are occasionally conflicting. For example, Schultz et al. [35] demonstrated that hemodynamic variables obtained for HR, MBP, and RPP by M-Pre were not significantly different compared to untreated control hearts in anesthetized rat hearts. However, another report by this group demonstrated that acute morphine treated-mice hearts showed significant improvement in recovery of RPP and left ventricular end-diastolic pressure [59].
Infarct size is considered the gold standard measurement for myocardial I/R injury measurement. Functional recovery after reperfusion also is a highly plausible end point and is considered a sensitive one by some authors [60]. However, contractile dysfunction reflects a combination of the effects of stunning and cell death. In addition, the intraventricular balloon may be a less sensitive method for measurement of functional recovery. In this regard, it is suspicious that hemodynamic parameters may accurately reflect cardioprotective effects. Tissue salvaged by an anti-infarct intervention leads to improvement of cardiac function; however, infarct size limitation does not always produce mechanical functional recovery of hearts after myocardial I/R injury. There is evidence that I-Pre and I-Post limit infarct size but do not reduce myocardial dysfunction [61,62]. A potent opioid such as fentanyl also reduces infarct size but does not protect against myocardial dysfunction in isolated rat hearts [63]. The discrepancy between infarct size limitation and cardiac function recovery may be explained by continued stunning, i,e. prolonged myocardial dysfunction in the histological absence of necrosis after I/R injury. It is proposed that stunning occurs in reversibly injured cells or via nonlethal injury to the epicardial border zones surrounding infarcted myocardium. Although cellular ATP levels or free radicals have been proposed to be important mediators for stunning [64], the exact mechanism responsible remains unclear. Another possible mechanism for myocardial stunning is an increase in the concentration of intracellular Ca2+ ([Ca2+]). The increase in [Ca2+] in myocardial cells likely explains the depressed ability of I/R hearts to generate contractile forces.
Go to :
Cross-talk between OPRs and adenosine receptors (ARs)
Interestingly, the OPR family is known to 'cross-talk' with other GPCRs families, such as adenosine receptors (ARs). Kato et al. [65] reported that the improvement of post-ischemic mechanical function by fentanyl administered before induction of ischemia was abolished by the selective A1AR antagonist 1,3-dipropyl-8-cyclopentylxanthine (DPCPX) in isolated rat hearts. They suggested the δ-OPR blockade inhibits A1ARs-mediated protection. Peart and Gross [66] also demonstrated that there is a functional coupling between OPRs and ARs. The cardioprotection afforded by the A1ARs agonist 2-chloro-cyclopentyladenosine or morphine was abolished by δ-OPRs antagonists BNTX or DPCPX, respectively, in anesthetized rat hearts. Taken together, it is likely that not only morphine but also remifentanil might 'cross talk' with ARs because these opioids activate δ-OPRs. Our recent unpublished data showed that the infarct-limiting effect by R-Post was significantly blocked by both naloxone and the nonspecific AR antagonist 8-(p-Sulfophenyl)theophylline, implying 'cross-talk' between R-Post and ARs in isolated rat hearts. Meanwhile, there also is evidence for 'cross-talk' between OPRs and the β-adrenergic receptors (ADRs) system in the heart [67,68]. For details on 'cross-talk' interactions between OPRs and β-ADRs in the heart, see the review by Pepe et al. [69].
Go to :
Morphine- and remifentanil-induced cardioprotection in humans
As a large number of studies performed in animals and cells strongly revealed that morphine- or remifentanil-induced preconditioning or postconditioning is cardioprotective, it should be determined if a similar effect is seen in the human heart. Growing evidence during recent years suggests that I-Pre and I-Post produce cardioprotection in animal as well as human hearts [70,71]. Tomai et al. [70] assessed whether the OPR antagonist naloxone abolishes adaptation to ischemia in humans during coronary angioplasty after repeated balloon inflations. In naloxone-treated patients, ST-segment change and cardiac pain severity during the second inflation were similar to those observed during the first inflation, whereas in placebo-treated patients, they were significantly lower. This finding reveals that I-Pre is mediated by OPRs in the human heart. Further more, I-Post also protects human hearts. I-Post performed immediately upon closing the lesion with guide wire and consisting of four cycles of 30 sec occlusion followed by 30 sec of reperfusion in patients undergoing primary coronary angioplasty resulted in reduction of infarct size, lower levels of creatine phosphokinase-myocardial band (CK-MB) fraction, and improved ejection fraction [71]. These beneficial effects persisted beyond 3 years. Bell et al. [72] reported that the expression of δ-OPRs in human atrial trabeculae isolated from patients undergoing coronary bypass grafting and δ-OPR stimulation mimicked I-Pre in human heart muscle in an hypoxic-reoxygenation model, which suggests a possible clinical use in myocardial ischemia treatment.
Although many studies with morphine or remifentanil during cardiac procedures have mainly focused on hemodynamics, anesthetic requirement or postoperative analgesics requirement, several interesting reports have been published on a possible pharmacological approach with morphine and remifentanil for the treatment of myocardial ischemia in clinical settings. Murphy and coworkers [73] administered 40 mg of morphine before cardiopulmonary bypass and evaluated the myocardial performance index with echocardiographic parameters intraoperatively. The myocardial performance index was improved by the administration of morphine prior to the ischemic insult of cardioplegic arrest. Pleym et al. [74] reported that 0.5 µg/kg/min remifentanil during coronary artery bypass graft in patients receiving remifentanil-isoflurane anesthesia reduced surgical stress but seemed to cause cardiac depression in the early postoperative phase. The patients who received supplemental remifentanil had a lower cardiac output, left ventricular stroke work index, and mixed venous oxygen saturation than placebo received patients. Recently, however, Xu et al. [75] reported that R-Pre decreased cardiac troponin I levels in patients undergoing off-pump coronary artery bypass surgery. R-Pre by 5 µg/kg of 10 min infusion after anesthetic induction markedly decreased the cardiac troponin I levels which is a high-specificity marker for cardiac injury after surgery compared to non-preconditioned patients. In addition, Wong et al. [76] reported that remifentanil administration 1 µg/kg bolus followed by a 0.5 µg/kg/min infusion for 30 min after anesthetic induction but before sternotomy yielded lower CK-MB levels in patients undergoing elective on-pump coronary bypass surgery.
Go to :
Summary
Morphine or remifentanil has been widely used perioperatively. A large number of in vitro and in vivo studies have demonstrated that morphine or remifentanil reliably offers cardioprotection. Both δ- and κ-ORs play a crucial role in morphine- or remifentanil-induced cardioprotection. The signaling pathways involved in the effects of these two opioids are currently being investigated; however, the cardioprotective effects of morphine or remifentanil in human hearts have not been fully examined yet. Further studies are needed to evaluate the effectiveness of morphine and remifentanil in a clinical setting.
Go to :
 XML Download
XML Download