

 PDF
PDF Citation
Citation Print
Print


Introduction
Although age-adjusted deaths resulting from atherodegenerative cardiovascular disease have decreased [1], the absolute number of deaths due to cardiovascular disease is projected to double over the next 50 years, primarily due to a disproportionate increase of the elderly in the population [2]. Considering that 40% of all deaths occur as a result of cardiovascular disease and that 65 million people in the United States of America (20% of the country's population) are affected by cardiovascular disease, the burden as a result of this disease on healthcare systems will escalate enormously [3]. Currently costing $350 billion a year in the United States, the economic impact is also staggering. Worldwide, including non-industrialized countries, cardiovascular disease is the leading cause of death with 17 million cardiovascular disease-associated deaths per year [4]. Thus, despite the introduction of statins and focus on risk modifications, there is an urgent need for the identification of targets and development of therapeutic biomolecules against atherosclerotic disease (Fig. 1).
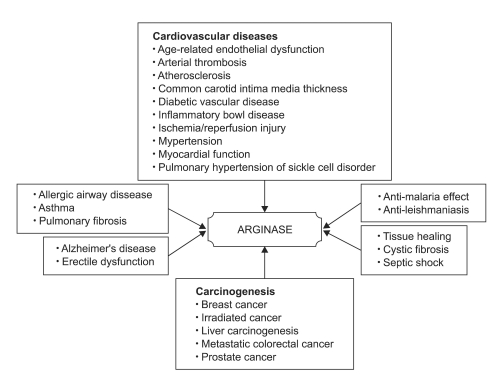 | Fig. 1Arginase inhibition and its therapeutic effects on diseases. Arginase is a prime therapeutic target protein predominantly emphasized by its role in asthma, cardiovascular and carcinogenic diseases. Furthermore, its application has been extended to preventing the progress of pulmonary fibrosis and erectile dysfunction. Focusing on cardiovascular diseases, atherosclerosis, hypertension, diabetic vascular disease and ischemia/reperfusion injury are the largest drug markets, occupying at least 25% of the total therapeutic market.
|
Go to : 
Endothelium Function and OxLDL in the Vascular System
The endothelium as a blood barrier plays a major role in the regulation of vascular homeostasis by maintaining the balance between vasoconstriction and vasodilation, as well as inhibiting and stimulating smooth muscle cell proliferation and migration. Endothelial dysfunction causing damage to the arterial wall is now considered an early marker for atherosclerosis, which induces attachment of monocytes to the endothelium, increases the permeability through the endothelial barrier and initiates oxidative modification of low-density lipoproteins. Modified LDL to oxidized LDL (OxLDL) particles are initially engulfed by macrophages, which utilize scavenger receptors on their surfaces, leading to foam cell formation. Formation of foam cells are indicative of the development of atherosclerosis [5]. Moreover, OxLDL appears to have potentially pro-atherogenic effects by inducing the expression of adhesion molecules on endothelial cells (ECs) [6], stimulating ECs apoptosis [7], inducing superoxide anion formation from NADPH oxidase, uncoupling endothelial nitric oxide synthase (eNOS) [8] and impairing endothelial nitric oxide (NO) formation, which is a major vasoprotective molecule released from the endothelium [9]. The responses to OxLDL in the endothelium are mediated by binding to the lectin-like OxLDL receptor-1 (LOX-1) [10], which activates multiple signaling cascades involving MAPK kinase [11,12], protein kinase C [13,14] and protein kinase B [15]. OxLDL increases caveolin I expression and interaction of NOS with this protein impairs NO formation [16]. Furthermore, OxLDL modulates PKC activity and expression and this family of kinases has been shown to acutely attenuate eNOS-dependent NO production and to promote eNOS uncoupling during prolonged exposure times [17]. In hypercholesterolemia, supplementation of L-arginine improves endothelial-dependent relaxation and NO production [18,19]. L-arginine also attenuates the development of atherosclerosis [20] and normalizes leukocytes adhesion [21] in hypercholesterolemia. Also hypercholesterolemia favors the accumulation of asymmetrical dimethyl-L-arginine (ADMA), a competitive inhibitor to L-arginine of eNOS in monkeys [22]. OxLDL increases the activity of the S-adenosylmethionine-dependent protein arginine methyltransferases [22], which generate ADMA from L-arginine. Moreover, OxLDL decreases the activity of the ADMA-degrading dimethylarginine dimethylaminohydrolase [23]. Therefore, OxLDL is a strong inducer of endothelial dysfunction, a state in which the endothelium produces superoxide anions rather than NO.
NO is derived from the substrate L-arginine by the action of eNOS in the presence of several co-factors including NADPH, BH4, FMN and FAD. eNOS is the enzyme constitutively expressed in the endothelium and its activity is regulated by both chemical agonists and mechanical forces (shear stress). Agonists increase intracellular Ca2+ concentrations through G-protein coupled receptors (GPCR) and increase activity through a Ca2+-calmodulin dependent mechanism. In contrast, shear stress activates NOS by phosphorylation through the Akt/PKB pathway which is Ca2+ independent [24,25]. Imbalance in the L-arginine/eNOS pathway to produce NO has been demonstrated to be critically important in a variety of pathophysiological conditions. Alternations in NO signaling have been shown in both animal models and human subjects with hypertension [26], diabetes [27], hypercholesterolemia [28,29], heart failure [29] and atherosclerosis [30].
Go to :
Arginase and Its Expression
Arginase is well demonstrated as an important enzyme in the last step of the urea cycle, which is the detoxification pathway for high concentration of ammonia by hydrolyzing L-arginine to ornithine and urea. There are two described isoforms, arginase I and II, encoded by different genes [31,32]. Arginase I has been known as the hepatic isoform, catalyzing the final step of the urea cycle, although recent studies demonstrate that arginase I can be induced by LPS, IL-13 and hypoxia in a wide variety of cells and tissues [33-37]. Arginase II has been referred to as the extrahepatic isoform and is inducible by a variety of factors: LPS, TNFα, interferon γ, 8-bromo-cGMP and hypoxia [34,35,38,39]. The extrahepatic isoform is widely distributed in numerous tissues. Additionally, arginase II provides ornithine for the synthesis of polyamines and L-proline, controlling cell proliferation, differentiation and repairing tissues following injury via ornithine decarboxylase [40,41]. Arginase II also contains a putative mitochondrial targeting sequence in the amino terminus and is thought to be confined primarily to the mitochondria [42,43]. It has recently been found that arginase reciprocally regulates NO production in ECs, that arginase is increased in the endothelium of ageing rats and that the inhibition or anti-sense knockdown of arginase I restores NO signaling and improved endothelial function in the vasculature of old rats [44,45]. In addition, the role of arginase in endothelial dysfunction has been extended to various cardiovascular disorders of animal models such as aging [44], ischemia-reperfusion-induced endothelial dysfunction [46], aortic coarctation hypertension [47], salt-induced hypertension and atherosclerosis [48]. In the penis, arginase II reciprocally regulates NO production and modulates erectile function. In the diabetic human corpus cavernosum, increased arginase II expression negatively regulates NO production and possibly contributes to erectile dysfunction [49].
Arginase II activity in OxLDL-stimulated human aortic endothelial cells (HAECs) is increased by two distinct mechanisms: (1) a novel early posttranslational event involving the release of the enzyme from microtubule association, resulting in activation; (2) transcriptional upregulation [50]. In thrombin-stimulated human umbilical vein endothelial cells, arginase II activity is increased through the Rho/ROCK pathway and the inhibitors Y-27632 and HA-1077 prevent arginase II activation without affection protein levels [51]. In LPS-treated macrophages, MAPK phosphatase-1 deficiency negatively regulates urea formation via greater expression of inducible NOS (iNOS) and NO production, although the overexpression or knockout of MKP-1 has no any substantial effect on the expression of arginases [52].
Go to :
Arginase and Reciprocal Regulation of Nitric Oxide
The three isoforms of NOS, eNOS, iNOS, and neuronal NOS (nNOS), produce NO from the substrate L-arginine, which is converted to L-ornithine and urea by the action of arginase. An emerging paradigm in NO biology is the concept that arginase, an enzyme that shares L-arginine as a substrate, reciprocally regulates NOS activity by competing for the substrate. Thus, arginase activity can effectively regulate NO-dependent processes by depleting the substrate pool available for NO biosynthesis (Fig. 2).
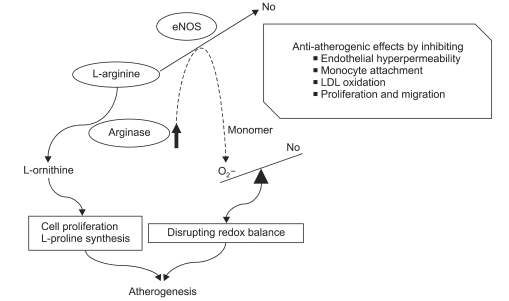 | Fig. 2Arginase roles in the atherogenic pathophysiology. Endothelial NOS catalyses L-arginine to L-citrulline and nitric oxide, which is an anti-atherogenic molecule produced in the endothelium. NO has been shown to have some protective effects by inhibiting endothelial hyperpermeability, monocyte attachment, the oxidative modification of low-density lipoproteins as well as the proliferation and migration of vascular smooth muscle cells. In endothelial cells (ECs), constitutively expressed arginase shares the common substrate, L-arginine. Arginase hydrolyzes L-arginine to urea and L-ornithine that is supplied for the synthesis of polyamines and L-proline. These are commonly used for cell proliferation and collagen synthesis which increases cross-linking in the extracellular matrix, leading to increases of vascular stiffness. Furthermore, increased arginase activity stimulates eNOS monomerization by depletion of their substrates and induces superoxide production by monomerized eNOS. Augmented superoxide production quickly disrupts the redox balance in ECs and leads to endothelial dysfunction. Therefore, arginase activation directly causes atherogenic vascular disease.
|
Arginase upregulation/activation impairs NO production by NOS isoforms and induces endothelial dysfunction associated with systemic hypertension [48,53]. Furthermore, compelling evidence suggests that arginase upregulation may aggravate pulmonary hypertension [54,55], a disease process known to be associated with impaired pulmonary vascular NO signaling. An increase in endothelial arginase activity contributes to the endothelial function and vascular stiffness associated with aging [44], although the isoform appears to be arginase I rather than arginase II. Increased arginase activity in aorta of ApoE-/- in mice is not correlated with an increase in arginase protein abundance. Arginase activation rather than an increase in protein abundance may be responsible for increased enzyme activity. In human aortic endothelial cells, the mechanism of increased arginase activity in response to OxLDL stimulation involves a rapid dissociation of the enzyme from the microtubular cytoskeletal structure, which appears to constrain its activity [50]. This is not reflected in vivo. This observed difference may be the result of species differences. The concept of species variability is supported by Hayashi et al. [56] who demonstrated that both arginase I and II abundances are increased in the vasculature of rabbits that are fed a high cholesterol diet and that arginase II abundance is modulated by estrogen administration. Being consistent with the vasculature, arginase II expressed specifically in myocyte constrains nNOS activity in the rat cardiac myocyte in an isoform-specific manner [57]. Arginase inhibition increases basal contractility in isolated myocytes by a nNOS-dependent mechanism, although the supplementation of L-arginine has no effect on myocyte NO production or myocyte contractility. Thus, this observation suggests that L-arginine pools are very tightly controlled for availability by the enzymes, iNOS and arginase II, in the myocyte.
There appears to be at least 3 pools of L-arginine that are spatially confined and regulated by different transporters and enzymes [58,59]. Thus, local concentrations of L-arginine in microdomains in which NOS and/or arginase might be located may be limiting for NOS isoforms. This is the concept of the L-arginine paradox that is found in the mammalian organism by which L-arginine concentrations by far exceed Km values of NOS. Therefore, additional L-arginine should not enhance NO formation. In vivo however, increasing L-arginine concentrations in plasma has been repeatedly shown to increase NO production.
With respect to enzymatic kinetics, the Km value of arginase is a range of 1-20 mM for L-arginine and that of NOS is 1-5 µM. This result implies that comparing the substrate competition between the enzymes is not effective. However, the catalysis rate of these enzymes has shown that arginase has a Vmax of 1400 µmol/min/mg, while NOS has a Vmax of 900 µmol/min/mg [60]. As cellular conditions contain much higher concentration of L-arginine than the Km value for NOS, a direct comparison of the Km value is not a reliable measurement for the catalytic properties of these enzymes [60].
Recently, Topal et al. [61], demonstrated using pharmaceutical techniques that three L-arginine pools exist : 1) a freely exchangeable pool (pool I) with extracelluar L-arginine that is regulated by the cationic transporter (CAT-1) and depleted by exchanging the pool with cationic amino acid lysine, 2) a nonfreely exchangeable pool (pool II) with extracellular L-arginine that cannot depleted by L-lysine, and 3) extracellular L-arginine pools (pool III) present in ECs and mitochondrial arginase II which modulates NO synthesis through a nonfreely exchangeable L-arginine pool. According to recent increasing paradigms, not freely exchangeable L-arginine pool II is composed of two cytosolic microdomains that pool IIA appears functions of the recycling of citrulline to arginine by combined reactions of arginosuccinate synthetase and arginosuccinate lyase [62]. The remaining L-arginine pool IIB reveals a property that cannot be depleted by neutral amino acids such as histidine.
The direct mechanisms are not well understood regarding how arginase activity can regulate or effect eNOS activity on a molecular level. A recent concept is that eNOS coupling is responsible for NO production and that eNOS uncoupling induces superoxide generation which is confirmed with OxLDL-stimulated ECs. Inhibition of arginase with S-(2-boronethyl)-L-cysteine (BEC) prevented the decrease in eNOS dimer bands. In addition, N (G)-nitro-L-arginine methyl ester (L-NAME) also prevented OxLDL-dependent decreases in dimer formation, and treatment of cells with L-arginine partially preserves eNOS dimer stability [63].
Go to :
ROS and the Vasculature
Under normal physiologic conditions, the use of oxygen by cells of aerobic organisms generates potentially deleterious reactive oxygen metabolites. A chronic state of oxidative damage exists in cells because of an imbalance between pro-oxidants and antioxidants. With regard to blood vessels, vascular production of reactive oxygen species (ROS) and the subsequent reaction and scavenging of NO is thought to be an important mechanism of vascular dysfunction in atherosclerosis [64] and hypertension [65]. There are three major enzymatic sources for vascular ROS and the mitochondrial electron transport chain (ETS): NAD(P)H oxidase, xanthine oxidase (XO), and NOS [66]. The NAD(P)H oxidase system has been under intense investigation ever since its discovery in the vasculature and its importance in atherosclerosis with impaired-NO dependent vasodilator function has been recognized. It was originally demonstrated that the NAD(P)H oxidase system was expressed in the smooth muscle and adventitial cells (fibroblasts) of the vascular wall and that function of the enzyme could be enhanced by angiotensin II [67]. The upstream signaling mechanisms by which angiotensin II enhances ROS production have recently been uncovered and appear to be protein kinase C mediated [68]. This signaling is recognized to be important in forms of hypertension in which the rennin angiotensin system is activated [69]. Endothelial XO may also contribute to increased oxidative stress and endothelial function associated with disease processes such as diabetes [70].
The role of NOS-3 in the generation of ROS is critically dependent on the bioavailability of L-arginine. In physiological conditions, L-arginine availability, at least when measured extracellularly, does not appear to be rate-limiting in the production of NO [71]. However, L-arginine concentrations in intracellular microdomains remain unknown. In fact, it has been shown that the administration of L-arginine restores, at least in part, the physiologic activity of NO in experimental hypercholesterolemia and hypertension [72]. The depletion of L-arginine leads to a derangement of the oxidase/reductase domains of NOS. Thus, NOS-3, when activated in an arginine depleted environment, is still able to receive electrons from NADPH and donate them to the substrate O2, resulting in one electron reduction to form O2-. It is established that O2- is produced by endothelial cells along with NO and that L-arginine availability may determine the ratio of O2- to NO produced. In addition, L-NAME can inhibit O2- generation by blocking NOS-3 activity through binding to the active site and decreasing electron flux to the heme site of the enzyme [73].
Go to :
NO, Vascular Stiffness and Atherosclerosis
The role of vascular stiffness in hypertension, in vascular disease associated with renal impairment and in the vascular changes that accrue with age is well documented [74-79]. Moreover, increased vascular stiffness represents a marker of increased vascular risk [79-83]. Although age is now recognized as the dominant cardiovascular risk factor, vascular stiffness is increasingly recognized as an even more powerful risk predictor because of the variability in vascular changes that occur with age and because vascular stiffness develops in non-age related conditions [84].
Atherosclerosis is primarily a disease that accrues with age. There is evidence that vascular stiffness is correlated with indices of advanced atherosclerotic disease in animal, human neurological and cardiac studies. It is unclear whether vascular stiffness perturbations reflect atherosclerosis or an age/pressure related phenomenon. However, mechanistically, certain studies have demonstrated a correlation between vascular stiffness and disturbances in lipid metabolism [85-87]. In its initial phases, atherosclerosis manifests itself by disturbances in EC function, NO bioavailability and oxidant stress, as demonstrated by impaired forearm [88] and coronary [89] endothelial-dependent vasodilatation. NO bioavailability has also been clearly demonstrated to modulate arterial stiffness [90]. However, vessel stiffness can also modulate endothelial cell signaling in response to luminal stimuli and the resulting NO release [91]. In clinical studies, high-resolution carotid ultrasonography has been used to obtain measurements of the thickness of the intima and media of the carotid arteries [92]. Previous studies have shown cross-sectional associations between common carotid artery intima-media thickness and cardiovascular risk factors, the prevalence of cardiovascular disease [93] and the involvement of other arterial beds with atherosclerosis [94]. Changes in commoncarotidartery intima-media thickness have also been adopted as a surrogate endpoint for determining the success of interventions that lowers the levels of low-density lipoprotein cholesterol [95]. Increases in intima-media thickness of the carotid artery, as measured non-invasively by ultrasonography, are directly associated with an increased risk of myocardial infarction and stroke in older adults without a history of cardiovascular diseases [93].
Go to :
Conclusions
Inhibition of endothelial arginase or deletion of the arginase II gene enhances NO production, restores endothelial function and aortic compliance, and reduces plaque burden. Arginase II represents a novel risk factor-independent target for the prevention and treatment of atherosclerotic vascular disease.
Go to :
 XML Download
XML Download