

 PDF
PDF Citation
Citation Print
Print


Introduction
Opioids, which have been widely used clinically as anesthetic adjuvants or for pain control, can protect against myocardial ischemic-reperfusion (I/R) injury [1,2]. It has been well known that the activation of opioid receptors by ischemic preconditioning (I-Pre) or opioid-induced pharmacological pretreatment provides cardioprotection against myocardial I/R injury [3,4]. However, because pretreatment is seldom possible in acute myocardial infarction, ischemic postconditioning (I-Post) or reperfusion-targeted pharmacological strategies recently has generated considerable interest. Surprisingly, only a few studies thus far have investigated the effect of opioid receptor agonists administered solely during reperfusion in myocardial I/R injury. In addition, these studies were mainly focused on non-specific opioid receptor agonists or a δ-opioid receptor (DOP) agonist [2,5].
The µ-, δ-, and κ-opioid receptors have been characterized pharmacologically. Binding and developmental studies have identified the presence of DOP and κ-opiod receptors (KOP) in the adult rat heart [6]. KOP are localized in both adult and neonatal rat hearts [7]. Although there is controversy regarding the role of KOP in myocardial I/R injury, a recent report demonstrated that activation of KOP by U50488H administered as a single bolus prior to reperfusion provided cardioprotection in intact rat hearts [8]. In an our recent report, we also showed that KOP activation early in the reperfusion period reduced myocardial infarction and improved cardiac function in isolated rat hearts [9]. However, the exact intracellular mechanism of cardioprotection by KOP activation is not well known.
Interestingly, the activation of reperfusion injury salvage kinase (RISK) signaling pathways, such as the p42/p44 extracellular signal-regulated kinases (ERK1/2) and pro-survival phosphatidylinositol-3-OH kinase (PI3K)-Akt cascades, at the time of reperfusion exerts cardioprotection against lethal myocardial reperfusion injury [9]. Cardioprotection by nonspecific opioid receptor agonists and a selective DOP agonist is known to occur via the PI3K-Akt signaling pathway [2], while the involvement of the ERK1/2 pathway is unclear.
The purpose of this study was to assess the role of the RISK signaling pathway, ERK1/2 and PI3K-Akt, in cardioprotection by exogenous KOP activation during reperfusion.
Materials and Methods
The experimental procedures and protocols used in this study were reviewed and approved by our institutional animal care and use committee.
Drugs and chemicals
U50488H (a standard selective KOP agonist), LY294002 (a potent and selective PI3K inhibitor), and PD98059 (an ERK1/2 specific inhibitor) were purchased from Tocris Bioscience (Ellisville, MO). 2,3,5-Triphenyltetrazolium chloride (TTC) was obtained from Sigma-Aldrich Chemical (St. Louis, MO). Fluorescent polymer microspheres were purchased from Duke Scientific (Palo Alto, CA). Antibodies against Akt and ERK1/2 were purchased from Cell Signaling Technology Inc. (Beverly, MA). Other chemicals were obtained from Sigma-Aldrich Chemical.
Langendorff isolated heart perfusion preparation
Male Sprague-Dawley rats (Korea Taconic Co, South Korea), weighing 280-330 gm were used for the experiments. They received 100 mg/kg of pentobarbital sodium and 300 IU of heparin intraperitoneally. Hearts were isolated and perfused with modified Krebs-Henseleit (KH) solution containing (in mM) 118.5 NaCl, 4.7 KCl, 1.2 MgSO4, 1.8 CaCl2, 24.8 NaHCO3, 1.2 KH2PO4, and 10 glucose. Regional ischemia and reperfusion were induced as previously described [10]. In brief, a snare was made at the level of the proximal length of left coronary artery. Regional ischemia was induced by pulling the snare and confirmed by regional cyanosis and a substantial decrease in left ventricular developed pressure. Reperfusion was started by releasing the snare.
Experimental protocol
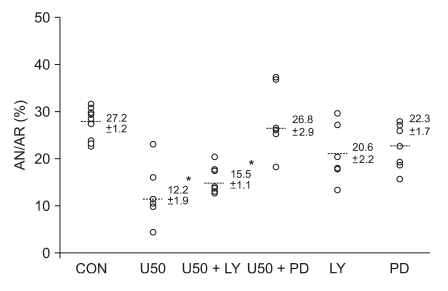
All hearts were subjected to 30 min of regional ischemia and 120 min of reperfusion, and each experimental group consisted of at least 7 hearts. In our previous report, the infarct volume as a percentage of ischemic volume in control hearts (CON) and 1 µM U50488H treated hearts (U50) were 27.2 ± 1.2% and 12.2 ± 1.9%, respectively (P < 0.05) [9] and these results were compared to the following treatment groups to determine whether the RISK signaling pathway would play a critical role in KOP agonist-induced cardioprotection (n = 7 for all groups): 1) U50 + LY; U50488H with 15 µM of LY294002, 2) U50 + PD; U50488H with 20 µM of PD98059, 3) LY; LY294002 alone, and 4) PD; PD98059 alone.
Exclusion criteria
We decided prospectively that any hearts with a heart rate < 250 beats/min, or coronary flow > 18 ml/min or < 8 ml/min at the end of stabilization would be excluded from the study. Hearts failing to develop left ventricular systolic pressure > 80 mmHg when the left ventricular end diastolic pressure was kept at 5-10 mmHg were also excluded. Any heart exhibiting arrhythmia during the stabilization period also was discarded.
Determination of area at risk and infarct size
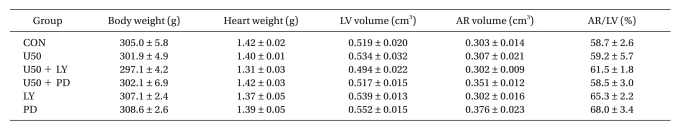
At the end of each experiment (after 2 h of reperfusion), the area at risk (AR) and area of necrosis (AN) were measured by TTC stain and quantified with Image Tool (UTHSCSA Image Tool, version 3.0) as described previously [12]. In brief, the coronary artery was re-occluded and fluorescent polymer microspheres were infused. The hearts were weighed, frozen, and cut into 2-mm slices. The slices were incubated in 1% TTC in sodium phosphate buffer 37℃ for 20 min. The slices were immersed in 10% formalin and then identified by illuminating the slices with UV light. The areas were converted into volumes by multiplying area by slice thickness. The AN volume was expressed as a percentage of the AR volume. All measurements were performed in a blinded fashion. There were no significant differences in body weight, heart weight, LV volume, AR volume, and AR/LV among the groups (Table 1).
Tissue lysate preparation and Western immunoblot analysis
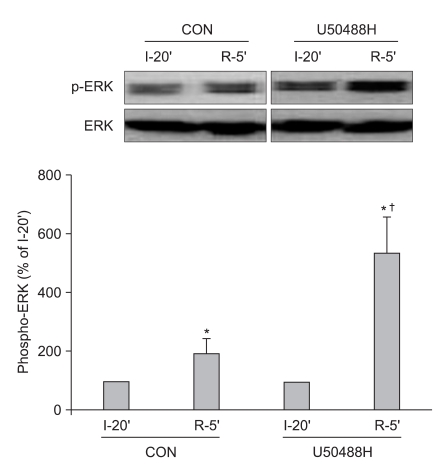
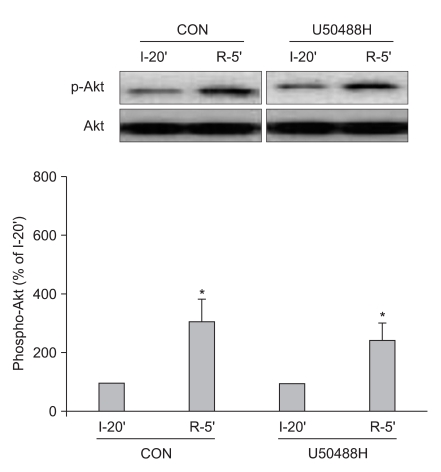
Additional hearts were randomly assigned into a control group (n = 6) and a U50488H treatment group (n = 6), and were subjected to 30 min of regional ischemia followed by 5 min of reperfusion. Myocardial samples were taken from the AR region at 10 min before and 5 min after reperfusion. Samples were homogenized in ice-cold lysis buffer. Equal amounts of protein (50 µg/lane) were loaded, subjected to electrophoresis on SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. Membranes were blocked with nonfat milk and then incubated with primary antibodies that recognize phosphorylation of Akt (protein kinase B) and ERK1/2 at 4℃ overnight. Bound primary antibodies were detected with a secondary anti-rabbit antibody and visualized by an enhanced chemiluminescence method. Data for total- and phospho-ERK1/2 represent the sum of the 42- and 44-kDa bands.
Statistical analysis
Data are presented as means ± SEM. Data analysis was performed with a personal computer statistical software package (SPSS for windows, Release 12.0; SPSS Inc, Chicago, IL). Data were analyzed using t-test and one-way analysis of variance (ANOVA) followed by a Bonferroni post-hoc testing. Differences were considered to be statistically significant when P values were less than 0.05.
Results
Infarct size studies with ERK1/2 and PI3K and inhibitors
As shown in Fig. 1, the ERK1/2 inhibitor PD98059 (26.8 ± 2.9%) completely blocked the anti-infarct effect of U50488H (P < 0.05 vs. U50). However, there was no significant difference in AR/AN between the U50488H treatment group and the PI3K inhibitor LY294002 treatment group (15.5 ± 1.1%, P > 0.05 vs. U50). LY294002 or PD98059 itself did not alter infarct size (20.6 ± 2.2% for LY and 22.3 ± 1.7% for PD, P > 0.05 vs. CON).
Western immunoblotting analysis of ERK1/2 and Akt
To determine the involvement of the RISK pathway in the cardioprotective effect of KOP activation, we further measured ERK1/2 (Thr202/Tyr204) and protein kinase B or Akt (Ser473) phosphorylation during reperfusion in perfused rat hearts. ERK1/2 phosphorylation significantly increased after 5 min of reperfusion compared to 10 min before reperfusion in both groups (189.3 ± 95.0% in CON and 534.1 ± 283.1% in U50, Fig. 2). Furthermore, U50488H-treated hearts had significant increases in ERK1/2 phosphorylation compared to control hearts (P < 0.05) immediately after reperfusion. Myocardial sample taken from AR at 5 min of reperfusion revealed an increase in Akt phosphorylation compared with samples taken 10 min before reperfusion (303.1 ± 76.2% in CON and 242.9 ± 46.2% in U50). However, there was no significant difference between the two groups (P < 0.05, Fig. 3).
Discussion
In the present study, the anti-infarct effect by U50488H was totally abrogated by the ERK1/2 inhibitor PD98059. However, the PI3K inhibitor LY294002 could not block the infarct reducing effect of U50488H. And these results were corroborated by the phosphylation status of ERK1/2 and Akt kinases using Western immunoblott analysis.
The anti-infarct effect of U50488H in our previous study [9] was comparable with those previously reported for the nonspecific opioid receptor agonist morphine or the δ1-opioid receptor agonist BW373U86 [2]. And, these results are consistent with a recent report by the Gross group [8], who showed that administration of either DOP or KOP agonists as a single bolus 5 min prior to reperfusion provided infarct size-sparing effects in intact rat hearts. Collectively, these data strongly suggest that not only DOP but also KOP stimulation during the reperfusion phase plays a role in preventing lethal myocardial reperfusion injury.
The signaling mechanisms of opioid receptor activation are unveiling and seem to be similar to those of I-Pre and I-Post. In the present study, the infarct size-sparing effect of KOP activation by U50488H was totally blocked by the ERK1/2 inhibitor PD98059 but not by the PI3K inhibitor LY294002, suggesting the involvement of ERK1/2 rather than PI3K-Akt signaling pathway in cardioprotection by KOP activation during reperfusion. This finding was also corroborated by direct measurement of ERK1/2 and Akt kinases using Western blots. Meanwhile, Peart and colleagues [13] reported that pretreatment with the PI3K inhibitor wortmannin abolished infarct reduction by U50488H in in vivo rat hearts. Of note, however, we measured infarct size using ERK1/2 and PI3K antagonists in KOP activation in conjunction with assessing the phosphorylation status. Our present study highly implicates the involvement of ERK1/2 but not PI3K-Akt in the cardioprotection achieved with KOP activation during reperfusion. We do not know the exact reason for the discrepancy of results between our and Peart's group, but the two pathways may also follow different spatio-temporal routes by KOP activation at early reperfusion, which should be defined in the future.
Meanwhile, the survival kinase cascades ERK1/2 and PI3K-Akt may exhibit 'cross-talk' such that inhibiting of one cascade may activate the other and vice versa [14]. However, it does not seem that cross-talk always occurs. For instance, a recent study stressed the pivotal role of ERK1/2 but not PI3K/Akt in I-Post mediated cardioprotection [15]. In this regard, the relative importance of the two kinases in mediating the protection by KOP activation remains controversial. The results from our study suggest that ERK1/2 but not PI3K/Akt is the predominant mediator of KOP-induced protection.
Interestingly, some drugs protect the myocardium against ischemic but not against reperfusion injury and vice versa [16]. Accordingly, when administering drugs to prevent myocardial I/R injury, it is important to know whether it is protective only when given before ischemia and/or when given after ischemia during reperfusion. In this regard, it has been well accepted that DOP stimulation is effective not only during the pre-ischemic period but also during the reperfusion period [17]. Although it seems that KOP is less influential in I-Post [18], there have been data to suggest that exogenous activation of KOP have an infarct-sparing effect in the myocardial I/R model. Peart and colleagues [19] reported that exogenously induced KOP activation by three different KOP agonists (U50488, ICI204448, and BRL52537) 10 min before the onset of ischemia reduced infarct size in an intact rat model of myocardial infarction. However, thus far there is little in scientific examining the effect of infarct-sparing associated with KOP activation by sustained drug administration during the reperfusion phase. Combined with previous reports of cardioprotection by KOP treatment before index ischemia [12,19], our result strongly suggest that KOP activation confers cardioprotection not only when given before ischemia but also when given after ischemia during the reperfusion phase.
In summary, our results suggest that sustained KOP activation targeting the reperfusion phase confers cardioprotection by the ERK1/2 subcellular signaling pathway but not the PI3K-Akt pathway. KOP activation during reperfusion, therefore, might be another cardioprotective mechanism.
 XML Download
XML Download