

 PDF
PDF Citation
Citation Print
Print


Introduction
Brain damage followed by cerebral infarction cause deformity/destruction of brain constantly even after reperfusion of cerebral blood flow was done. The opening of blocked blood flow as one of treatments for cerebral infarction can cause 2nd damage by damaging reperfusion which produces active oxygen and free radicals [1]. So, even numbers of studies about medicines or treatments to reduce destruction of brain cell have been done, but there have been yet no surely verified treatments. Ischemic preconditioning by means of repeated treatments of intermittent block of blood flow and reopening suggested by Murry et al. [2], was known as a treatment to operate strong intrinsic suppressive mechanism for prevention damage of ischemia organs, but since it can be limitedly applied only under situation that ischemia may be predicted theoretically, it has limitation in applying that it should be done clinically in extremely limited situation. Recently, it was reported first by Zhao et al. [3] that ischemic postconditioning performed on heart of experiment dog, that is, repeated treatments of intermittent block of blood flow and reopening for short time direct after ischemia, has similar prevention effect to ischemic preconditioning for myocardial necrosis after reperfusion. Relating to cerebral blood flow, Zhao at al. [4] presented that intermittent block of blood flow in reperfusion result in significant reduction of brain damage after brain ischemia/reperfusion locally in Rat, and suggested treatment method for cerebral infarction. However, even though numbers of studies have been performed, exact suppressive mechanism has not been yet found. It is significant meaning in that Postconditioning can be applied after ischemia compared to preconditioning.
Heat shock protein (Hsp) is a protein which can make cells to cope with stress that cause harmful stimulus and environmental damage [5], and also restrain deformity of protein and reduce inflammatory response, and have cytoprotective effects for reducing cell necrosis and apoptosis [6]. Hsp70 has been recognized to be increased relating to brain ischemia [7], and lot of efforts has been made to reduce brain damage by increasing expression of Hsp [8]. Regarding relevance between Hsp70 and neuroprotective effect, there are increasingly reported recently [9,10], but unfortunately mostly studied in only limited specific model and also didn't clarify clearly not only about causal relationship with ischemic postconditioning but also about proper cycle and time to appear neuroprotective effect and reperfusion method. The purpose of this study is to review whether effect of brain protection is or not, degree of the effect, and influence on expression of Hsp 70 which are appeared by hemispatial ischemical postconditioning in local brain ischemia/reperfusion which make reperfusion by inducing ischemia phase after blocking middle cerebral artery (MCA) of Rat.
Go to : 
Materials and Methods
Ischemia/reperfusion in middle cerebral artery (MCA)
All animal experiment has been performed under permit of animal experiment ethics committee and also has complied with animal care policy of committee. Using 20 male rats of Sprague Dawley weighing 250-300 gr, they was fed by water and prey normally at the interval of 12 hours until experiment day in breeding room equipped with dimmer. Experiment animals were classified at random into Controls group and ischemic postconditioning group having 10 rats respectively. Experimental rats were injected zoletil 12.5 mg and xylazine 3mg into abdominal cavity, and then removing fur around neck and fixing them in supine position at animal guides, putting clinical thermometer inside rectum, keeping their body heat 36.5℃ using heating pad, and keep their spontaneous breathing properly during operation.
Operation method of middle cerebral artery occlusion referred to previously reported method [11]. Rods in 400 µm thick were made by heating 4-0 nylon monofilament (Johnson & Johnson, USA). Performing midline incision at cervical region, and then after desquamating ambient thyroid, nerves, and fascia under dissecting microscope, common carotid artery in both side were found and left common carotid artery was exposed to branch point between internal carotid artery and external carotid artery. Distal portion of external carotid artery and internal carotid artery was tied up loosely and after holding common carotid artery with curved microvascular clip, and then incise external carotid artery, 4-0 nylon monofilament rod was inserted inside internal carotid artery around 20-21 mm depth until feeling of resistance.
After operation of middle cerebral artery of control group, ischemia happened for 60 minutes, and so, put forceps on left common carotid artery for that time, and then after 60 minutes, pull 4-0 nylon monofilament rod outside so as not to break external carotid artery, take off clip in common carotid artery, and finally reperfuse middle cerebral artery. For postconditioning group, after inducing them for ischemia in same environment with control group, and then pull nylon thread out to branch point of common carotid artery and release clip in left common carotid artery for 30 seconds to reperfuse and to induce ischemia again. Reperfusion and Ischemia were repeated in 3 times in same way. Experiment animals were moved to breeding room after coming out of anesthetic followed by removal of nylon thread and suture the skin.
Measurement of neurological score
After 24 hours since brain damage, neurological score of rat was measured. In neurological scores, score 0 was in case of normal, score 1 was in case if forelegs were bent on hang up rat by tail, score 2 was in case if gripping force of forelegs was reduced, score 3 was in case that it can move to any direction but when was pulled up by tail, it turn to opposite side, and lastly, score 4 was in case if it turn to opposite side even in remaining quiet with nothing [12].
Measurement of infarction areas and observation of cerebral edema
After measuring neurological score and anesthetizing using zoletil 12.5 mg and xylazine 3 mg, decapitation was done and brain was extracted. Extracted brain was also amputated along observing surface towards frontal lobe from branch point of middle cerebral artery at the interval of 2mm, and then amputated tissue dyed in 2% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma, USA) which was dissolved in saline solution at 37℃ for 30 minutes, and was fixed at 4℃ by phosphate-buffered formaline. Infarct area was not dyed and was shown in white similar to existing brain tissue, while normal tissue became dyed in red. Total area of infarct area was analyzed by image analysis system (Image I software, NIH, USA), and ratio of infarction was converted total area of ipsilateral cerebral hemisphere divide by infracted area [13], cerebral edema formation ratio of infracted area was calculated by area ratio of both cerebral hemisphere inside same amputated surface [14].
Immunohistochemical analysis
After making paraffin cake in 4 µm thick from infracted area of each groups and brain tissue of object in which cerebral edema was observed 24 hours later from brain damage, deparaffinizing it using xylen and dehydrate it by ethanol and distilled water step by step, and then activity of caspase-3 and activity of Hsp 70 was investigated in immunohistochemical method.
To investigate activity of caspase-3, commercialized kit (Sigma Statin® Cleaved Caspase-3 (Asp 175) IHC kit, cell signaling technology, USA) was used. After dropping blocking solution on tissue cake, it was treated with primary antibody and negative control, and then it was washed out with phosphate buffered saline (PBS) 24 hours later. Biotinylated secondary antibody was dropped on slide and then 30 minutes later after that, it was washed out with PBS, and AB reagent which was made by blending reagent A and B was dropped on this. After dropping substrate-chromagen which was made by blending ready-made substrate with distilled water on slide, waited until it become reddish brown. And then contrast dye was performed using hematoxylin. Since that, it was dehydrated using xylene and it was fixed with fixatives.
Immunohistochemical analysis of Hsp70 was performed by diluting Hsp70 antibody (HSP70 Antibody #4872, cell signaling technology, USA) in 1 : 50. For Antigen unmasking, tissue slide was soaked into 10 mM of sodium citrate buffer (pH 6.0) and was boiled for 10 minutes and then was cooled down. Tissue slide is applied to 3% hydrogen peroxide for 10 minutes and washed out by distilled water and tris buffered saline (TBS)/0.1% Tween-20 (TBS), and then blocking solution which was made by blending normal goat serum with TBST was dropped on it to be applied to room temperature for 1 hour, and treating primary antibody to it and applying it for 24 hours at 4℃. Since then, it was washed out by TBST and AB reagent was dropped on it. After substrate-chromagen was dropped on slide, contrast dye was performed using hematoxylin. Since then, it was dehydrated by ethanol and xylem and was fixed, and permanent mounting medium was dropped on it and was covered by cover glass.
After number of active cell of caspase-3 and Hsp70 was observed and measured in optical microscope of 400 times magnification selecting 3 places by every 1 mm2 at hemispheric frontoparietal cortex (boundary zone of local ischemia) and caudoputamen (central zone of local ischemia) in which local ischemia/reperfusion were performed, the median number was taken.
Statistical processing
Result value was presented in the median number (a quartile). Comparison of morality rate and score of neurological score between the control group and the experiment group was conducted in Chi-square and Fisher's exact test, and area of infarction and edema, number of active cell of caspase-3 and Hsp70 were analyzed using Wilcoxon rank sum test or Mann-whitney rank sum test. Sigma stat (version 3.1, Stat software, USA) was used for statistic analysis, and it was found to be significant when P value was less than 0.05.
Go to :
Results
Mortality rate
There was no died object in both the control group and the ischemic postconditioning group until 24 hours later after brain damage.
Neurological score
Neurological score which had been observed 24 hours later after brain damage showed score 0 in 5 rats among 10 rats and score 1 in other 5 rats of the control group, while showing same score distribution in ischemic postconditioning. No differences were found between two groups.
Comparison of formation degree of cerebral infarction and edema
There was no difference between the control group and the ischemic postconditioning group in formation rate of cerebral infarction and edema (Fig. 1)
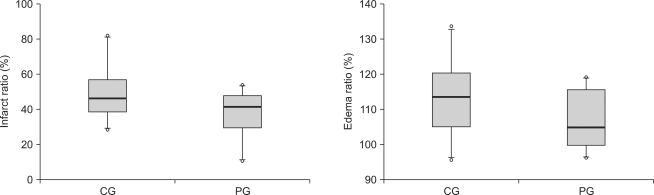 | Fig. 1Infarct ratios and edema ratios, measured at 24 hours after cerebral ischemia/reperfusion injury. Infarct ratios are calculated as the percentage of the size of the infarcted tissue area/ipsilateral hemisphere. Edema ratios are calculated as the percentage of the size of the ischemic hemisphere/contralateral normal hemisphere. The boxes show interquatile ranges; thick lines are medians and error bars are 10th and 90th percentiles. There are no significant differences between the groups. ◦: Extreme cases over 3 times of each box value. CG: control group, PG: ischemic postconditioning group.
|
Comparison of number of active cell of Caspase-3 and Hsp70
There was no difference in number of active cells in Caspase-3 at both frontoparietal cortex and caudoputamen between the control group and the ischemic postconditioning group 24 hours later since brain damage (Fig. 2, 3, 4).
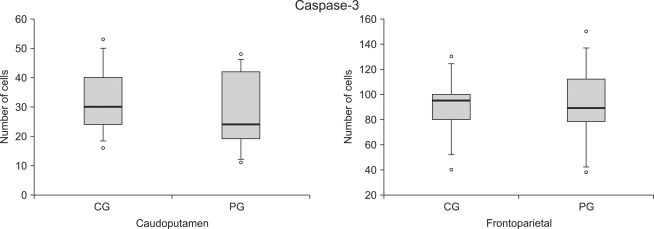 | Fig. 2The number of caspase-3 reactive cells at the caudoputamen and the frontoparietal cortex following cerebral ischemia/reperfusion injury. The boxes are interquatile ranges; thick lines are medians and error bars are 10th and 90th percentiles. There are no significant differences between the groups. ◦: Extreme cases over 3 times of each box value. CG: control group, PG: ischemic postconditioning group.
|
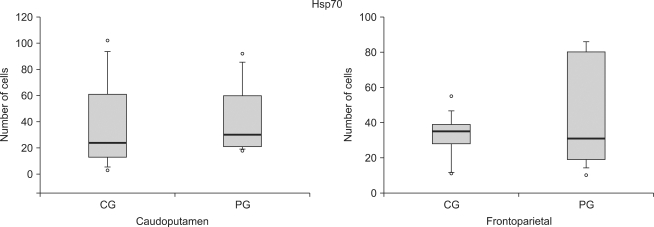 | Fig. 3The number of Hsp70 reactive cells at the caudoputamen and the frontoparietal cortex following cerebral ischemia/reperfusion injury. The boxes are interquatile ranges; thick lines are medians and error bars are 10th and 90th percentiles. There are no significant differences between the groups. ◦: Extreme cases over 3 times of each box value. CG: control group, PG: ischemic postconditioning group.
|
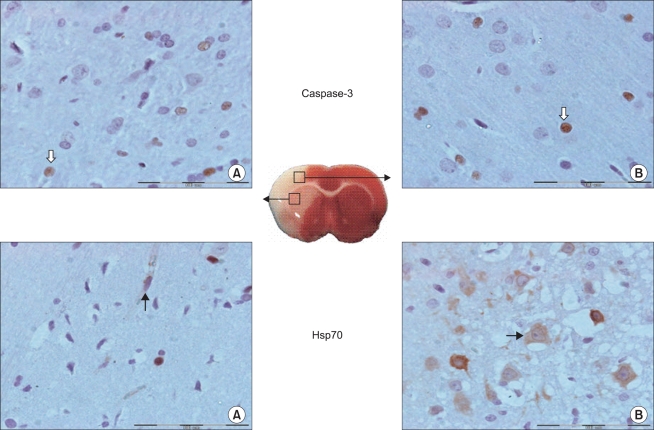 | Fig. 4Representative microscopic images of caspase-3 and Hsp70 reactive cells following cerebral ischemia/reperfusion injury. Cells are measured in the two regions of the ischemic hemisphere: in the caudoputamen (A) and the frontoparietal cortex (B). Caspase-3 reactive (white arrow) and Hsp70 reactive (black arrow) cells are shown. The boxes show 400× magnification. Scale bar = 100 µm in the insets.
|
Go to :
Discussion
Ischemic postconditioning, which was performed in way of that reperfusion of ipsilateral common carotid artery for 30 seconds after ischemia of unilateral middle cerebral artery for 60 minutes and blockage for 10 seconds in consecutively 3 times, didn't reduce early brain damage after local brain ischemia/reperfusion. Also in comparison of apoptosis and expression of Hsp70 through immunohistochemical dye, there was no significant difference from the control group. In particular, number of active cell of caspase-3 and Hsp70 were not different from the control group in both central area and ambient area of ischemia respectively. It meant that neuroprotective effect of ischemic postconditioning could not be expected in early brain damage, since it may not induce reduction of nerve damage by apoptosis and increase of Hsp70.
The period of reperfusion after ischemia is very important in pathophsiological mechanism in which damage was caused. It has been known that in the mechanism of brain damage due to cerebral ischemia and reperfusion, there are the damage of ischemia/reperfusion [15], mechanism of cell necrosis [16], the mechanism of apoptosis related to cytochrome C and caspase-3 [17], the mechanism of mitogen-activated protein kinase [18], the mechanism of protein kinase C [19], the mechanism of Akt [20]. Ischemic postconditioning, which was regarded as one of mechanisms to have something with cerebral protective effects among mechanisms due to damage of cerebral ischemia/reperfusion, has been known to reduce death of nerve cell by restraining apoptosis. However, according to previous researches, in a partial reperfusion model in which firstly middle cerebral artery was permanently blocked and then both sided common carotid artery was temporarily blocked, and then reperfuse it, when ischemic postconditioning had been performed after blocking for 15 minutes and 30 minutes, reduction effect of infarction area was big, and superoxide product, which was measured 30 minutes after reperfusion, was reduced, and 2 days after damage of cerebral ischemia/reperfusion, it also reduced increase of TUNEL positive cells around ambient ischemia, and also ischemic postconditioning reduced oxidative stress material which was caused during reperfusion and reduced damage by restraining apoptosis [4]. Xing et al. [10] reported when postconditioning, in a way of repeatedly reperfusion for 30 seconds and blockage for 30 seconds in consecutive 6 times after temporary blocking of middle cerebral artery for 60 minutes in rat, was conducted, Bcl-2 which was antiapoptosis protein was increased, and also movement of Bax and cytochrome C from mitochondrial to cytoplasm was reduced. Consequently, postconditioning is recognized to restrain mechanism of apoptosis of cortical neuronal cell due to damage of ischemia/reperfusion. In this study, Infarction area and edema was measured 24 hours after brain damage in group which was applied by ischemic postconditioning, and when counting number of active cell of caspase-3 in caudoputamen section and ambient frontoparietal, no significant difference was found in the control group respectively. Therefore, ischemic postconditioning was recognized to have little neuroprotective effects in early brain damage and could not restrain effectively mechanism of Apoptosis. However, depending on model of experimental animals and damage, postconditioning effects varied and exact time and cycle of reperfusion/block in postconditioning to have cerebral protective effect has not clarified yet [4,10]. Reperfusion for 30 seconds and then block for 10 seconds in a cycle of 3 times was applied in this study. But, even though same cycle of reperfusion/block with previous methods reported was applied [4], as a method was used in way which middle carotid artery was blocked temporarily not permanently and then reperfused it and on reperfusing middle carotid artery, only one side of common carotid artery not both side carotid artery was tied up and off by forceps, partial movement of blood flow through collateral circulation, which was possible to be existed in circle of Willis or proximal part of middle carotid artery, could not be perfectly controlled. With this reason, more effective ischemia and reperfusion were recognized not to be induced. Zhao [21] reported that to induce ischemic damage on one sided middle carotid artery and treat ischemic postconditioning on both sided common carotid artery and both sided middle carotid artery is difficult to expect cerebral protective effect of ischemic postconditioning due to more severe brain damage than permanent ischemic damage of middle carotid artery. To expect neuroprotective effect by ischemic postconditioning directly after reperfusion, it was important to decide duration time and number of cycle for reperfusion and block respectively, and when reperfusion will be conducted [22]. It was generally known that proper observation period for damage after local cerebral ischemia is 2-24 hours, but, in case of light and temporary local ischemia damage, expression might be delayed because of mechanism of Apoptosis differently from damage of permanent or severe ischemia for above 90 minutes [23]. So, if it will be studied for longer observation period, significant findings can be obtained. In addition, since other paths except caspase-dependent path among paths of Apoptosis can affect [24], if research for quantification is carried out side by side to quantify major proteins in caspase-dependent and independent path such as cytochrome C, Bcl-2, and Bax protein and apoptosis inducing factor, relationship with Apoptosis should be more clearly clarified.
It was known that Hsp70 was increased responding to stress such as ischemia [25], acted not only as chaperone but also restrained the course of cell-death such as cell necrosis or Apoptosis and control inflammatory response [6,26]. Overexpression of Hsp70 could come into neuroprotective effect in cultured neuronal cell [27], and also could reduce brain damage even in experimental model for stroke of rat [28]. There is little research to report that among cell mechanism to reduce damage due to cerebral ischemia and reperfusion, postconditioning was related to expression of Hsp, but there is some research to report that Hsp was increased relating to pharmacological postconditioning and ischemic postconditioning [9,10]. O'sullivan et al. [9] reported that after inducing rat to cerebral infarction and shock, when postconditioning was carried out using diazoxide which was channel opener of mitochondria Kapt, Hsp25 and Hsp70 were increased. Xing et al. [10] reported that after blocking of middle carotid artery of rat, and then ischemic postconditioning was carried out on reperfusion, materials relating to mechanism of Apoptosis and the expression of Hsp70 were measured. As a result, Hsp expression was increased after postconditioning, and ischemic postconditioning restrained Apoptosis. Consequently, it had neuroprotective effect to reduce damage of local cerebral ischemia/reperfusion. But unfortunately in this study, influence of ischemic postconditioning on expression of Hsp70 was not clarified as above. This corresponded in some degree with this study which resulted in that brain damage was not reduced after ischemic postconditioning. And, it also related to result that Apoptosis was not reduced. It can be said that authors' expectation that number of Hsp70 active cell will be increased in group who was treated by ischemic postconditioning is wrong. However, in this study, indirect method which calculated number of Hsp70 active cell was used. Therefore, by applying quantitative method such as Western blot to measure direct activity, it is needed to grasp actually how much extent the expression of Hsp70 was arose.
It was thought that the reason why there was few difference of degree in cerebral infarction between the ischemic postconditioning group and the control group was that there had only marginal influence of ischemic postconditioning on protection of blood vessel in nerves. Damage on permeable barrier of micro vessel was appeared within 1-2 hours after blocking of middle cerebral artery, and then Transudate was arisen and destruction of blood brain barrier was started. Vascular nerve had played important role in ischemic cerebral damage [29]. However, the influence of death of endothelial cell in vascular nerve on cerebral edema due to damage of cerebral ischemia have not been clearly clarified yet, and no study have been done yet about the influence of neuroprotective effect in ischemic postconditioning on structure of vascular nerve. The study about this issue is highly required.
It was thought that the reason why there was no big difference in neurological score was that degree of damage was not big and serious so as to cause functional disability. Neurological test methods, which was used in this study, was well known that it reflected clearly relationship between area of cerebral infarction and functional disability but in case of small infarction area, it had no relationship with neurological deficits [30].
In conclusion, to apply ischemic postconditioning after nerve damage due to temporary local cerebral ischemia/reperfusion in Rat had not improved area of cerebral infarction, reduction of edema, and neurological improvement. Therefore, it can be recognized as having no neuroprotective effect in early damage. Regarding mechanism relating to apoptosis and Hsp70, further study is highly required.
Go to :
 XML Download
XML Download