

 PDF
PDF Citation
Citation Print
Print


Introduction
Gabapentin, 1-(aminomethyl)cyclohexane acetic acid, is a structural analogue of the neurotransmitter γ-aminobutyric acid (GABA) [1]. Gabapentin was introduced in 1993 as an adjuvant anticonvulsant drug for the treatment of refractory partial seizures [2]. Experimental stuedies have shown that gabapentin can reduce neuronal injury in the setting of cerebral ischemia [3,4].
Although gabapentin was originally modeled based on the structure of GABA, the drug does not modulate GABA receptor function in the manner of conventional GABAergic drugs, and gabapentin is inactive at GABA receptors. The drug has been shown to increase GABA content in neuronal tissues and to bind to the α2δ subunit of calcium channels [5]; the biological effects of gabapentin are largely attributable to the selective inhibition of high-voltage-activated calcium channels containing the α2δ-1 subunit [6]. It has been postulated that neuroprotective effects of gabapentin might be related to changes in glutamate synthesis and metabolism [1]. A recent study showed that the in vivo neuroprotection associated with gabapentin was directly attributable to antiseizure effects and not to a direct cellular neuroprotective mechanism [4], but details of the cellular model of gabapentin action remain to be determined.
This study was conducted to determine whether gabapentin pretreatment altered the expression of heat shock protein 70 and reduced acute phase neuronal injury in rats subjected to transient focal cerebral ischemia/reperfusion.
Go to : 
Materials and Methods
Preparation of animals
This study was performed with the approval of our Institutional Animal Care and Use Committee, and in accordance with NIH guidelines for the care and use of laboratory animals. Adult male Sprague-Dawley rats weighing 260-300 g were used. The rats were housed at constant humidity and temperature with a 12-hour light/dark cycle and free access to food and water. Rats were randomized into four groups (10 rats per group); these were a saline-treated control group (CG), and groups receiving 0.1, 0.5, and 5 mg/kg of gabapentin pretreatment (0.1 GG, 0.5 GG, and 5 GG).
Rats were anesthetized with zoletil 12.5 mg and xylazine 3 mg intraperitoneally and respiration was maintained spontaneously. Core body temperature was monitored using a rectal probe and was maintained at 37.0 ± 0.5℃ employing a heating pad (Homeothermic Blanket System, Harvard Apparatus Inc., USA) during all experiments. Indwelling i.v. cannulas (BD Angiocath Plus™, 0.75 in, 0.7 × 19 mm, Becton Dickinson, Korea) were placed into the left femoral veins of all animals to permit drug delivery.
Focal cerebral ischemia was induced by occluding the left middle cerebral artery (MCA) as described previously [7]. In brief, under an operating microscope, the left carotid bifurcation was exposed through a midline cervical incision. The external carotid was ligated distally and the branches were coagulated. An arteriotomy was made in the external carotid allowing the introduction of a 4-0 surgical nylon monofilament suture (Johnson & Johnson, Belgium) with the tip rounded by heating near a flame. The filament was gently advanced into the internal carotid artery 18-20 mm past the carotid bifurcation until a slight resistance was felt, thereby occluding the origin of the MCA. Sixty minutes after MCA occlusion (MCAO), reperfusion was allowed by withdrawal of the suture thread until the tip cleared the internal carotid artery.
Rats of the gabapentin pretreatment groups received 0.1, 0.5, or 5 mg/kg gabapentin (Sigma-Aldrich, USA) dissolved in physiologic saline 20 minutes before the onset of middle cerebral artery occlusion. Control rats received 100 µl physiologic saline.
Evaluation of neurological deficits
The neurologic status of each rat was carefully evaluated in surviving animals at 1 and 3 days after MCAO. Neurological defects were determined using a modified Bederson's scoring system [8]. Each rat was held gently by the tail, suspended 1 m above the floor, and forelimb flexion observed. Control rats extended both forelimbs toward the floor. Rats that extended both forelimbs in this manner and that had no other neurological deficits were assigned grade 0. Rats with any amount of consistent forelimb flexion contralateral to the injured hemisphere and no other abnormality were graded 1. Rats were placed on a large sheet of soft, plastic-coated paper that could be gripped firmly by the claws. With the tail held by hand, a gentle lateral pressure was applied behind the shoulder of each rat until the forelimbs slid several inches. Rats with reduced resistance to lateral push toward the paretic side were graded 2. Rats were then allowed to move about freely and were observed for circling behavior. Rats that circled toward the paretic side when the tail was pulled were graded 3, and rats that circled toward the paretic side consistently without any stimulation were graded 4.
Analysis of cerebral infarct
After neurological evaluation, rats were randomly chosen from each group, given an overdose of thiopental sodium, and decapitated. The brain was quickly removed and cut into 2-mm-thick coronal blocks. Brain slices were immersed in a 2% (w/v) solution of 2,3,5-triphenyltetrazolium chloride in normal saline at 37℃ for 30 minutes and next fixed in 10% (v/v) phosphate-buffered formalin at 4℃. The stained brain slices were photographed using a CCD video camera and the size of each infarct was calculated using an Image Analysis System (Image J software, National Institutes of Health, USA). Infarct area was expressed as the percentage of infarcted area with reference to the area of the ipsilateral ischemic hemisphere and hemispheric swelling was calculated as the percentage increase in the size of the ipsilateral hemisphere compared with the contralateral intact hemisphere [9].
Imunohistochemical analysis
Brain tissues were sectioned at a thickness of 4 µm and cells showing caspase-3 and HSP70 activity were counted.
A commercial kit (Sigma Statin® Cleaved Caspase-3 (Asp175) IHC kit, Cell Signaling Technology, USA) was used to assess the activity of caspase-3 following the manufacturer's instructions. Deparaffinized and rehydrated sections were immersed in 0.01 M sodium citrate buffer (pH 6.0), heated at a temperature less than the boiling point for 10 minutes, and cooled for 30 minutes to permit antigen unmasking. Next, 1-2 drops of peroxidase quench solution were applied to each slide. After washing in distilled water and phosphate-buffered saline (PBS), 1-3 drops of blocking solution was applied followed by 1-3 drops of primary antibody or negative control solution. After 24 hours incubation, each slide was washed in PBS and biotinylated secondary antibody was applied for 30 minutes. Each slide was washed in PBS, premixed AB reagent was added, and a substrate-chromagen mixture was next applied. Staining was monitored until sections turned red-brown in color. After counterstaining with hematoxylin, slides were dehydrated in ethanol, treated with a permanent mounting medium, and a coverslip applied.
Anti-Hsp70 antibody (HSP70 Antibody #4872, Cell Signaling Technology, USA), at a 1 : 50 dilution, was used to assess Hsp70 levels as described by the manufacturer. Deparaffinized and rehydrated sections were immersed in 10 mM sodium citrate buffer (pH 6.0) and heated at a temperature less than boiling for 10 minutes before cooling for 30 minutes to achieve antigen unmasking. Next, slides were washed in distilled water three times. Slides were incubated in 3% (v/v) hydrogen peroxide solution for 10 minutes, washed in distilled water and Tris-buffered saline (TBS)/0.1% (v/v) Tween-20 (TBST), and 100-400 µL blocking solution was applied for 1 hour at room temperature; 100-400 µL primary antibody was next added. Each slide was then incubated at 4℃ overnight, washed three times in TBST, and biotinylated secondary antibody diluted in TBST, according to the manufacturer's recommendation, was next applied for 30 minutes at room temperature. After washing in TBST, AB reagent was added, followed by a substrate-chromagen mixture, and staining was monitored until color development was evident. After counterstaining in hematoxylin, each slide was dehydrated in ethanol, and a permanent mounting medium and a coverslip were applied.
Caspase-3- and Hsp70-reactive cells were counted under a light microscope in high-power fields (×400) in three random and nonoverlapping 1 mm2-sized regions in the caudoputamen (ischemic core) and the fronto-parietal cortex (the penumbra).
Statistical analysis
All data are expressed as medians with interquartile ranges. The chi-squared and Fisher's exact tests were applied for intergroup comparisons of death rate and neurological scoring. Differences between infarcted areas, edema ratio, and numbers of caspase-3- and Hsp70-reactive cells between groups were analyzed with the Kruskal-Wallis test. A P value less than 0.05 was considered significant.
Go to :
Results
Twenty-four hours after transient MCA occlusion/reperfusion, a well-defined pale and collapsed infarct area was seen in the cerebral hemisphere (Fig. 1). Neurologic scores were not significantly different between groups.
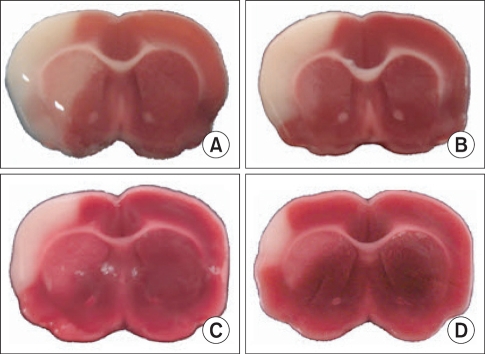 | Fig. 1Middle cerebral artery occlusion/reperfusion-induced brain injuries are shown. Representative images of 2,3,5-triphenyltetrazolium chloride-stained sections from saline- and gabapentin-treated animals. (A: Saline treated control group, B: 0.1 mg/kg gabapentin group, C: 0.5 mg/kg gabapentin group, D: 5 mg/kg gabapentin group). A well-defined pale and collapsed area, considered to be an infarction, is seen in the left hemisphere.
|
The mean infarct area in rats pretreated with 5 mg/kg gabapentin was significantly less than in saline-treated rats (P < 0.05), but was not significantly smaller than the areas in 0.1 mg/kg and 0.5 mg/kg gabapentin-treated animals. The edema ratios of gabapentin-pretreated rats were significantly reduced compared with those of saline-treated rats (P < 0.05) (Fig. 2).
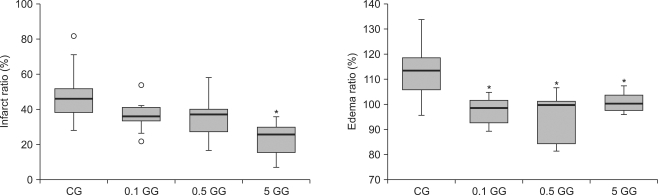 | Fig. 2Infarct ratios and edema ratios measured 24 hours after cerebral ischemia/reperfusion injury. An infarct ratio was calculated as the percentage of the size of the infarcted tissue area with reference to the size of the ipsilateral hemisphere. Each edema ratio was calculated as the percentage of the size of the ischemic hemisphere with reference to that of the contralateral normal hemisphere. The boxes show interquartile ranges; the thick lines are medians and the error bars are the 10th and 90th percentiles. *P < 0.05 compared with the saline group. ◦: Extreme examples of over three-fold each box value. CG: saline treated control group, 0.1 GG: 0.1 mg/kg gabapentin group, 0.5 GG: 0.5 mg/kg gabapentin group, 5 GG: 5 mg/kg gabapentin group.
|
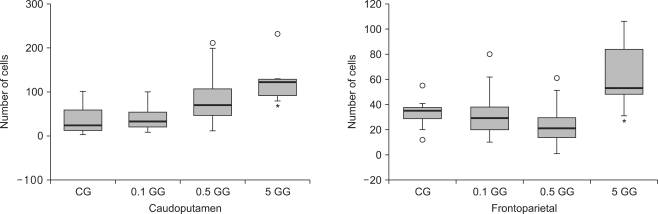
The number of caspase-3 reactive cells in gabapentin-treated rats was not significantly different from numbers in saline-treated rats in either the caudoputamen or fronto-parietal cortex. There were more Hsp70-expressing cells in the 5 mg/kg gabapentin group than in control animals, both in the caudoputamen and fronto-parietal cortex (P < 0.05) (Fig. 3-5).
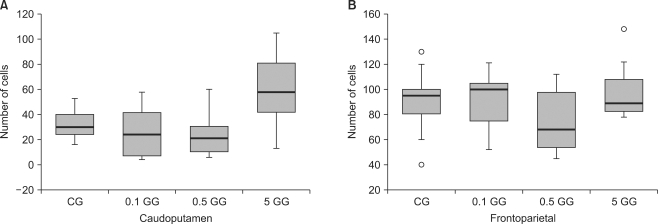 | Fig. 3The number of caspase 3-reactive cells in the caudoputamen and the fronto-parietal cortex following cerebral ischemia/reperfusion injury. The boxes are interquartile ranges; thick lines are medians, and error bars are the 10th and 90th percentiles. There were no significant differences among the groups. ◦: Extreme examples over three-fold each box value. CG: saline treated control group, 0.1 GG: 0.1 mg/kg gabapentin group, 0.5 GG: 0.5 mg/kg gabapentin group, 5 GG: 5 mg/kg gabapentin group.
|
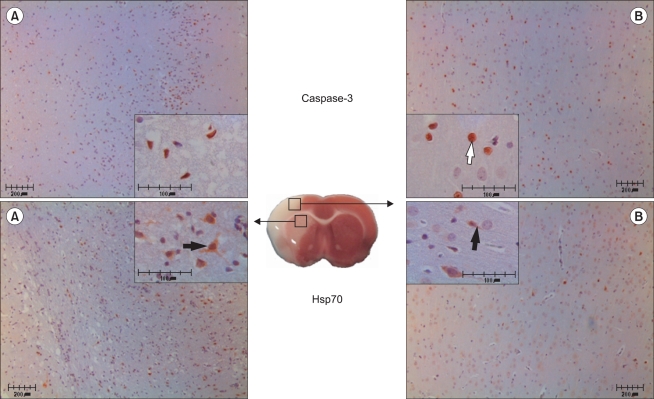 | Fig. 5Histopathologic findings in the caudoputamen (A) and the fronto-parietal cortex (B) following cerebral ischemia/reperfusion injury. Representative microscopic images of caspase-3-(white arrow) and Hsp70-reactive (black arrow) cells in a saline-treated rat. Large and small boxes show 100× and 400× magnifications, respectively. Scale bars = 200 µm and 100 µm in insets. P < 0.05 compared with the saline-treated group.
|
Go to :
Discussion
After transient MCA occlusion/reperfusion, gabapentin, particularly at 5 mg/kg, reduced infarct volume and cerebral edema in all treated groups. Moreover, the number of cells showing Hsp70 reactivity increased in the 5 mg/kg gabapentin groups. However, gabapentin did not cause a decrease in the number of caspase-3 reactive cells. These results indicate that gabapentin pretreatment can mitigate early brain injury induced by MCA occlusion/reperfusion in the rat and that the mechanism of neuroprotection is related to expression of Hsp70.
Although the cellular mechanisms of gabapentin pharmacological action are not well understood, several hypotheses have been proposed, suggesting that different properties account for the anticonvulsant, antinociceptive, anxiolytic, and neuroprotective activities of gabapentin in animal models [1,10,11]. A possible pharmacologic effect of gabapentin is modulation of calcium currents by selective drug binding to the auxiliary subunit of voltage-dependent calcium channels [6], and it has been postulated that the neuroprotective effects of gabapentin might arise from changes in glutamate metabolism [1]. As the ischemic pathway overlaps significantly with seizure processes, anticonvulsants have been proposed as possible neuroprotective agents [12]. It has been reported that high-dose gabapentin significantly decreased acute seizures frequency and reduced brain atrophy in immature mice receiving unilateral carotid artery ligation [3]. Williams and colleagues [4] reported that gabapentin significantly reduced infarct volume and that the neuroprotective effect of gabapentin was attributable to an antiseizure property, and not to an effect on cellular mechanisms. In the present study we administered gabapentin intravenously 20 minutes before brain injury induced by transient MCA occlusion/reperfusion and found a significant reduction in infracted area in the 5 mg/kg gabapentin group. The cerebral brain edema ratio decreased significantly in all gabapentin-treated animals compared with the control group. The present study showed that neuroprotective effects were seen after administration of lower doses of gabapentin, compared with doses reported previously [13,14] and it may therefore be postulated that cellular mechanisms other than antiseizure efficacy, such as overexpression of Hsp70 and prevention of cerebral edema, are relevant to drug action.
Another explanation for the neuroprotective effects of lower gabapentin doses might involve the route of administration. It may be expected that effective therapeutic blood levels in the central nervous system can be more rapidly and reliably achieved by intravenous injection rather than oral, intraperitoneal, or subcutaneous administration.
The extent to which cell death during stroke involves apoptosis, necrosis, or a combination of both, remains controversial [15,16]. Of the potential pathways for implementation of apoptosis, activation of caspases, a family of cysteine aspartate proteases, is involved both in the signaling cascade and in execution of apoptosis by degradation of key intracellular proteins [17]. In the present study, the number of caspase-3-reactive cells in gabapentin-pretreatment groups was not significantly different from that in the saline-treated control group. This indicates that delayed neuronal cell death triggered by apoptosis cannot be prevented by gabapentin.
The number of cells showing Hsp70 activity in the 5 mg/kg gabapentin-treated group was significantly greater than in control animals. When it is considered that both infarct size and cerebral edema level were significantly decreased in the same group, this indicates that the neuroprotection afforded by gabapentin is related to overexpression of Hsp70. This protein, a member of an evolutionarily conserved constitutive and/or inducible protein family acting as molecular chaperones, is by far the best-studied of the ATP-dependent chaperone proteins and is induced by specific types of stress, including heat, oxidative challenge, protein cross-linking, and exposure to heavy metals [18]. Although the mechanism by which Hsp70 affords neuroprotection from cerebral ischemia was initially attributed to chaperone activity, recent work has suggested that Hsp70 may also directly interfere with cell death pathways such as apoptosis and necrosis and may also modulate inflammation [19-23]. However, in the present study, the number of caspase-3-reactive cells in gabapentin-pretreatment groups did not differ from levels in control animals.
Hemispheric brain edema in rats of gabapentin-pretreatment groups was less than seen in control rats. In addition to neuronal cell death, cerebral microvessels are targeted in cerebral disease states including focal ischemia, and this may derive from the rapid microvascular response to such ischemia, indicating that cerebral microvessels and their neighboring neurons may be associated in a neurovascular unit [24]. However, to the best of our knowledge, no previous study has investigated the effects of gabapentin on cerebral edema associated with endothelial disruption after ischemic brain injury in an animal model. Therefore, reduced edema in gabapentin-treated rats might be explained by an inhibitory effect of gabapentin on N-methyl-D-aspartate-induced excitotoxicity and effects on ion channels such as the voltage-gated sodium and calcium channels, rather than a protective drug effect on the blood-brain barrier [25,26].
In conclusion, gabapentin pretreatment significantly reduced infarct volume and cerebral edema after focal cerebral ischemia induced by transient MCA occlusion/reperfusion in the rat and increased the number of cells showing Hsp70 reactivity. These results indicate that gabapentin may have a neuroprotective effect and can reduce early neuronal injury caused by focal cerebral ischemia/reperfusion, and this may be mediated by expression of Hsp70. However, gabapentin pretreatment did not prevent the caspase-dependent apoptosis. As gabapentin is drug used in clinics, these results strongly support a potential use in clinical therapy for ischemic cerebrovascular disease.
Go to :
 XML Download
XML Download