

 PDF
PDF Citation
Citation Print
Print


Introduction
Spinal cord ischemic injury is a progressive neurological disorder including paraplegia which may be primarily associated with initial mechanical injury to spinal cord and secondarily additional neuronal cell death caused by substances released from cells in response to interruption of spinal cord blood flow [1]. Paraplegia has been reported at incidences ranging from 4.6 to 21% following aneurysm surgeries of the thoraco-abdominal aorta [2]. In spite of various strategies proposed or used to reduce the incidence of paraplegia by spinal cord ischemia injury after surgery, clinically effective methods developed for spinal cord protection are not yet well established.
Several studies have suggested that ischemic preconditioning represents protective effects in many organs including heart, brain, and spinal cord [3-6]. Ischemic preconditioning consists of two phases; an early phase wanes over several hours, but the protective effect reappears hours after the stimulus and lasts for several days (delayed phase) [3,4]. Isoflurane, a commonly used volatile anesthetic, has been demonstrated to be neuroprotective against brain and spinal cord ischemia [5,7-10]. It has been reported that isoflurane-induced delayed preconditioning reduces spinal cord ischemic injury via activation of mitochondrial adenosine triphosphate-dependent potassium channel, release of free radical, or nuclear factor κB (NF-κB) expression [7,8,11]. Although several cellular mechanisms on delayed preconditioning effect of isoflurane have been proposed, the exact signaling and cellular mechanisms in spinal cord protection by isoflurane-induced preconditioning are not fully understood. Of them, role of nitric oxide (NO), the main focus of our attention, on the effect of isoflurane-induced preconditioning after spinal cord ischemia have still not been elucidated. Actually, it has been well known that NO generated by inducible nitric oxide synthase (iNOS) has both neuroprotective and neurotoxic effects in the central nervous system (CNS) [12]. Mostly, iNOS has been found to be a major factor to initiation of the CNS inflammatory or degenerative conditions through the production of excessive NO [12]. However, recent study showed that enhanced expression of iNOS activity in the cardiac ischemia reduced infarct size [3] and another study also showed that NO plays an important role on cardiac preconditioning [13]. Related to the spinal cord injury, NO induced by iNOS was implicated to be neurotoxic in the subacute phase after spinal cord ischemia [14].
Numerous studies have demonstrated that NF-κB plays a central role in the regulation of many genes responsible for the generation of mediators or proteins in inflammation [15]. NF-κB activation which produces neuroprotective effect of isoflurane-induced delayed preconditioning was closely linked with initiation of iNOS transcription [12].
We hypothesized that NF-κB and iNOS might be in a sequence of isoflurane-induced neuroprotection. The present study was designed to investigate the possible relationship between the pre-ischemic isoflurane exposure on spinal cord ischemia and iNOS expression and the role of NF-κB by using iNOS-specific antibody and pyrrolidinedithio carbamate (PDTC), NF-κB inhibitor in the ventral horn of spinal cord in rats.
Go to : 
Materials and Methods
Male Sprague-Dawley rats weighing 200 ± 10 g (8 weeks in age) were obtained from a commercial breeder (Charles River Technology, Orient Co., Seoul, Korea) for the experiment. The experimental procedures were performed in accordance with the animal care guidelines of National Institute of Health (NIH) and the Korean Academy of Medical Sciences. The animals were housed under the controlled temperature conditions (20 ± 2℃) and lighting (from 07:00 h to 19:00 h) conditions and were supplied with food and water ad libitum.
The animals were divided into five groups (n = 6 in each group): the sham group, the control group, the PDTC-treated group, the isoflurane-treated group, the PDTC/isoflurane-treated group. The PDTC-treated groups were administrated with intraperitoneal injection of 2% 100 mg/kg PDTC (Sigma, St. Louis, MO, USA) at 1 h before operation and at 24 h and 48 h after reperfusion. The rats in the isoflurane groups received 30 min inhalation of 2.8% (2.0 MAC) isoflurane (Minrad Inc, Main St. Buffalo, NY, USA) at 24 h before spinal cord ischemia as a method of isoflurane preconditioning. The experimental design is illustrated in Fig. 1.
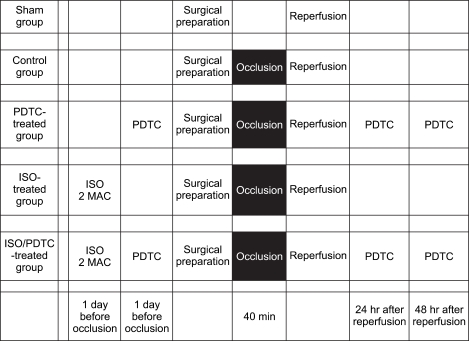
Prolonged ischemia was induced based on the method previously described by Zhou et al. [16] Briefly, the animals were first anesthetized by intramuscular injection of Zoletil 50® (50 mg/kg; Virbac Laboratories, Carros, Frances). The peritoneal cavity was then exposed through a vertical midline incision at the upper abdomen. The abdominal aorta was exposed and isolated. Abdominal aorta occlusion was induced by placement of a microvascular clamp around the aorta at the level just distal to the left renal artery. Successful arterial occlusion was indicated by the immediate cessation of pulse in the distal artery and pale appearance of the hind paws of the animals. The microvascular clamp was released after 40 min and the abdominal incision was closed in two layers with silk suture. In the sham-operated rats, the same surgical procedure was carried out, but the abdominal aorta was not clamped. All animals were allowed to recover in each cage at 25℃ after operation.
For spinal cord tissue preparation, the animals were sacrificed 72 h after reperfusion. In brief, the animals were fully anesthetized with Zoletil 50® (25 mg/kg, i.m.), transcardially perfused with 50 mM phosphate-buffered saline (PBS), and then fixed with a freshly prepared solution consisting of 4% paraformaldehyde (PFA) in 100 mM phosphate buffer (PB, pH 7.4). Following perfusion, the entire lumbar segments of the spinal cord (L1-6) were dissected and removed. The spinal tissues were postfixed for 24 h in the same fixative and then finally kept in the phosphate buffer (pH 7.4) containing 15% sucrose overnight at 4℃. Transverse sections of 20 µm thickness for histochemistry and immunohistochemistry were made using a freezing microtome (Leica, Nussloch, Germany). Spinal cords in all groups were excised for histopathological evaluation at 72 h after reperfusion.
iNOS immunohistochemistry was performed based on the method previously described by Zhou et al. [16]. Briefly, free-floating sections of the spinal cord were first immersed in phosphate-buffered saline containing 0.2% Triton X-100 (Sigma) (PBST, pH 7.4). The sections were quenched with 0.3% H2O2 for 20 min to remove endogenous peroxidase. After being washed with PBST, the sections were incubated in 4% normal horse serum (Vector Laboratories, Burlingame, CA, USA) for 1 h to block nonspecific binding. Afterward, the sections were incubated overnight at room temperature (25℃) with iNOS-specific mouse monoclonal antibody (Transduction Laboratories, Lexington, KY, USA) at a concentration of 1 : 500 with 0.1 M PBS (pH 7.4). The sections were then washed three times with PBST and incubated for 1 h with a biotinylated anti-mouse secondary antibody (1 : 200; Vector Laboratories). Subsequent antibody detection was carried out using VECTASTAIN® ABC Kit (Vector Laboratories). For visualization, the sections were incubated in 50 mM Tris-HCl (pH 7.6) containing 0.02% 3,3'-diaminobenzidine (DAB; Sigma) and 0.03% H2O2 for 5 min, and the sections were finally mounted onto gelatin-coated slides. The slides were air-dried at room temperature, and coverslipped with Permount® (Fisher Scientific, Fair Lawn, NJ, USA).
The numbers of motor neurons stained with iNOS-positive cells in the ventral gray matter region were surveyed and counted hemilaterally in 10 sections at L3-L5 segments of spinal cord using a light microscope (Olympus, Tokyo, Japan). Differences were determined by one-way ANOVA followed by Duncan post-hoc test and considered significant for P < 0.05. The results were expressed as the mean ± standard error of the mean.
Go to :
Results
Photomicrographs of iNOS-positive cells in the ventral horn of spinal cord at 72 h after reperfusion are presented in Fig. 2. The number of iNOS-positive cells counted in the ventral gray matter region was 7.50 ± 0.75/section in the sham group, the control group was 3.70 ± 0.59/section, the PDTC-treated group was 3.12 ± 0.52/section, the isoflurane-treated group was 5.45 ± 0.43/section, and the PDTC/isoflurane-treated group was 2.42 ± 0.21/section (Fig. 2).
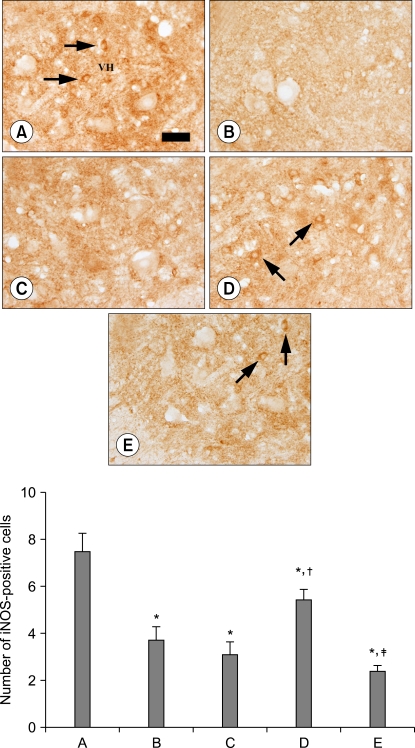 | Fig. 2Preconditioning effect of isoflurane on the number of iNOS-positive cells after spinal cord ischemia. Upper: photomicrographs of iNOS-positive cells in the ventral horn of spinal cord at 72 h after reperfusion. The arrows indicate iNOS-positive cells. (A) sham group, (B) control group, (C) PDTC-treated group, (D) isoflurane-treated group, (E) PDTC/isoflurane-treated group. Scale bar represents 25 µm. VH: ventral horn. Lower: the number of iNOS-positive cells in the ventral horn of spinal cord. The data are presented as the mean ± standard error of the mean. (A) Sham group, (B) control group, (C) PDTC-treated group, (D) isoflurane-treated group, (E) PDTC/isoflurane-treated group. PDTC: pyrrolidinedithio carbamate . *P < 0.05 compared to the sham group. †P < 0.05 compared to the control group. ‡P < 0.05 compared to the isoflurane-treated group.
|
The present results showed that spinal cord ischemia significantly decreased the iNOS expression in all groups compared to the sham group (P < 0.05) and a pre-treatment with PDTC reduced the iNOS expression compared to the control group, but not significantly. Preconditioning of isoflurane significantly increased the iNOS expression compared to the control group (P < 0.05), whereas pretreatment both with PDTC and isoflurane significantly decreased the iNOS expression compared to the isoflurane-treated group (P < 0.05).
Go to :
Discussion
The present study demonstrated that pre-ischemic isoflurane exposure increases iNOS expression in the ventral horn of spinal cord in rats. This mechanism of action can be clinically important to surgical patients because isoflurane is present before and during intraoperative ischemic insults.
A lot of studies have suggested that preconditioning with pharmacological agent and volatile anesthetics improves histological outcomes after spinal cord ischemia [5,15,17]. In particular, isoflurane preconditioning is reported to enhance the survival rate of neuronal cells after ischemia injury [5,7,8]. Sang et al. [8] showed that repeated inhalation with 1 MAC isoflurane each day for 5 consecutive days induced a delayed phase of ischemic tolerance to spinal cord ischemia in rabbits after 24 and 48 h but not 72 h.
The major finding of the present study is that isoflurane-induced delayed preconditioning significantly increased iNOS expression and this increase of iNOS expression was partly regulated by NF-κB. It has been well known that iNOS is the major contributor to initiate inflammatory and degenerative conditions through the production of excessive NO in the CNS [12]. Excessive production of iNOS has been thought to play an important role in the extension of neuronal injury during the reperfusion period following the ischemic insult [18,19]. Zhou et al. also reported that induction of iNOS in the motor neurons of the ventral horn may be involved in the selective and delayed neuronal death in the spinal cord induced by ischemic insult [16]. Moreover, NO generated by the iNOS of macrophage, neurons, oligdentrocytes, and astrocytes causes apoptotic cell death after spinal cord ischemia [20,21]. Therefore, these studies insist that neuroprotective mechanism of isoflurane-induced delayed preconditioning reduces neuronal apoptosis induced by iNOS.
In contrast, some other studies are consistent with our present results. NO acts as second messenger and controls important cellular processes by regulation of expression of certain proteins [12]. Several studies have suggested that the late phase of ischemic preconditioning for protection against cardiac ischemia is associated with selective upregulation of iNOS [22]. In addition, previous studies have shown that increase of iNOS, especially induced by isoflurane, could act positively in various organs. Sjakste et al. [23] showed that isoflurane increases iNOS in rat brain cortex and cerebellum, whereas Maeda et al. [24] reported that isoflurane did not inhibit interleukin 1β (IL-1β)-induced expression of iNOS and iNOS mRNA in the rat aorta. Recently, Wakeno-Takahashi et al. [3] suggested that isoflurane induces second window of preconditioning through upregulation of iNOS from 24 h to 72 h after cardiac ischemia. Inhibition of NO production caused marked increase in hind limb motor disturbance [14]. Peri-ischemic iNOS inhibition did not ameliorate neurologic and histopathologic outcome after transient spinal cord ischemia [17]. Together with previous studies, the present results carefully suggest the possibility that NO induced by iNOS may act as an important mediator of delayed preconditioning with isoflurane for the protective effect against spinal cord ischemia as observed in cardioprotection.
The possible mechanisms of iNOS induction have been proposed. Previous studies have reported that NO modulates important physiological processes in cells by regulation of expressions of certain proteins such as NF-κB [12], and NF-κB is essential for the induction of iNOS by lipopolysaccharide in murine macrophages [25]. Also, activated NF-κB increases the potent inducers of iNOS such as TNF-α and IL-1β [26]. Moreover, in the late phase of ischemic preconditioning, NO generated by the eNOS and acting via the formation of reactive oxygen species, activates the epsilon isoform of protein kinase C, which activates the Src family of protein tyrosine kinases and NF-κB with resultant upregulation of the iNOS gene and protein expression. iNOS and other cardioprotective proteins, including cyclooxygenase-2 and aldose reductase, confer resistance to subsequent ischemic stress [27].
The present results showed that induction of iNOS by isoflurane-induced delayed preconditioning after spinal cord ischemia is reduced by PDTC, a selective NF-κB inhibitor. Related to the spinal cord ischemia, several studies have reported that activation of NF-κB in spinal cord ischemia is pretty important to the CNS pathophysiology and inhibition of NF-κB prevents lung injury induced by intestinal ischemia and reperfusion [28,29]. In contrast, NF-κB activation in astrocytes leads to the synthesis of neurotrophins such as nerve growth factor and brain-derived neurotrophic factor essential for neuronal survival [30]. Although the function of NF-κB after spinal cord ischemia is still controversial, the present result suggested that NF-κB may be one of the mediators for modulating of iNOS during isoflurane-induced delayed preconditioning.
Here in this study, we have demonstrated that that pre-ischemic isoflurane exposure significantly increased iNOS expression via a pathway modulated by NF-κB in the delayed phase of spinal cord ischemia. We also expect isoflurane-induced iNOS activation may be beneficial to neurologic outcome after spinal cord ischemia.
Go to :
 XML Download
XML Download