

 PDF
PDF Citation
Citation Print
Print


Pulmonary hypertension (hereinafter PHT), which has numerous causes, results from increased pulmonary resistance and constriction of pulmonary vessels. Clinical treatment of pulmonary hypertension is difficult, and the disease entity can indirectly trigger right ventricular failure, which can lead to death. Studies conducted in the 1980s revealed that nitric oxide (NO) is related to the etiology of PHT, and various drugs for PHT treatment with minimal NO side-effects have recently been developed. Here, we discuss clinically erroneous prognoses of the pathophysiology of PHT, the mechanism of NO treatment, its actual use in practice, its toxicity, and other drug therapies used for the treatment of PHT.
Pathophysiology of pulmonary hypertension
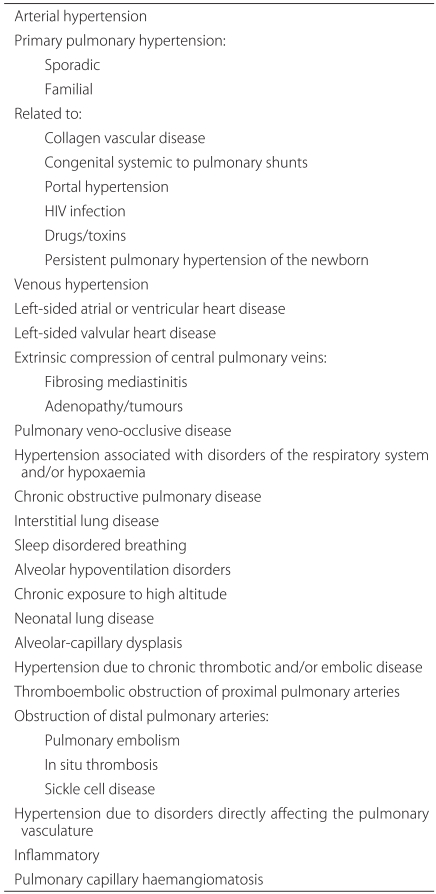
PHT is the occurrence of pulmonary resistance accompanied by elevated pulmonary arterial pressure. There are primary and secondary causes of PHT. Secondary PHT is characterized by prolonged vasoconstriction caused by different acute, chronic pulmonary or cardiac disorders. This condition creates a structural abnormality in the blood vessel base that is accompanied by hypoxia and an inflammatory response, which eventually leads to elevated pulmonary resistance. It is important to note that secondary PHT also causes the original disorder to worsen (Table 1) [1,2]. Primary PHT is defined as a pulmonary arterial pressure greater than 25 mm Hg at rest or greater than 30 mm Hg during exercise. If not treated, the life span of an individual with PHT is about 2.8 years and the 5-year survival rate is only around 34%. The ultimate cause of death as a result of primary PHT is right ventricular dysplasia and right ventricular failure. Although quite rare, primary PHT is most commonly found in Caucasians, younger individuals and women. In addition, primary PHT is occasionally dominantly-inherited [3,4].
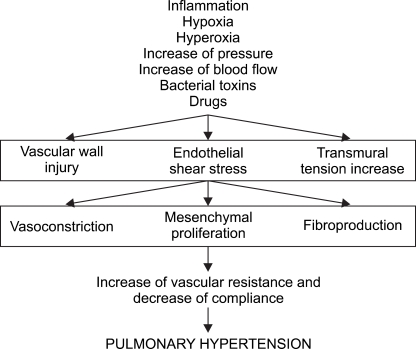
Elevated pulmonary resistance during PHT occurs due to in creased vascular tone and structural remodeling of the peripheral pulmonary arteries. During remodeling, vascular smooth muscles undergo hypertrophy and proliferate, which results in the quantity of vascular connective tissue increasing. As the endothelial cells are impacted, the blood vessel dia meters eventually constrict and pulmonary resistance in creases. Normal pulmonary circulation is characterized by a high blood flow rate, low resistance and low pressure. Pulmonary resistance is controlled by vascular smooth muscle cells and oxygen tension inside the alveoli, K+ channel activity and many vasoactive mediators. However, when pulmonary blood flow increases abnormally, pulmonary blood vessels are continuously strained from the pressure and the blood vessel walls are damaged. As a result, mesons such as angiotensin II, endothelin-1, 5-hydroxytryptamine and inflammatory cytokine are produced. In cells, Ca++ and protein kinase-C act as mediators, thicken blood vessel walls and induce the inside walls to grow, thereby causing remodeling [5-8]. Pulmonary resistance also increases in response to decreases in the number of pulmonary vessels per segment. Such changes begin with peripheral arteriole vascular smooth muscle before the capillaries.
As the formation of abnormal blood vessels progresses, control of the vascular tone decreases and the blood vessels con strict. This causes hypoxia, which then causes hypoxic pulmonary vasoconstriction. As this process repeats, it eventually leads to abnormalities in the endothelium. In other words, structural abnormalities cause functional abnormalities (Fig. 1) [9]. The cause of acute and chronic hypoxia in PHT differs slightly. Specifically, hypoxic pulmonary contraction is the cause of acute hypoxia, whereas elevated pulmonary resistance due to structural remodeling is the main cause of chronic hypoxia [10].
Nitric oxide in pulmonary hypertension
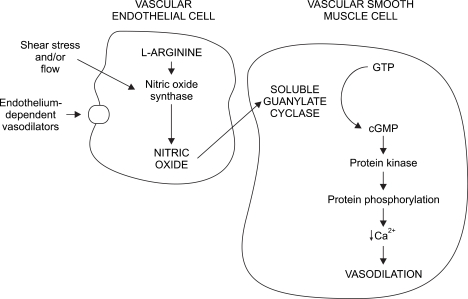
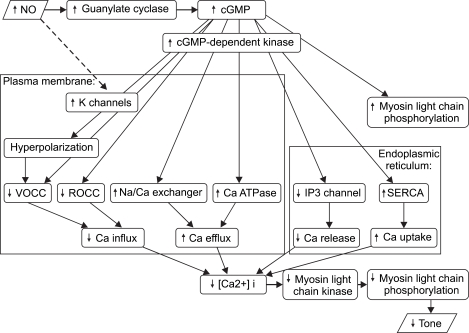
NO has a low molecular weight, is oleophilic and is a very fast-reacting endogenous free radical. In vascular endothelial cells, NO is produced by NO synthases (NOS). The precursor to NO is the terminal guanidine nitrogen of L-arginine, and its formation also requires nicotinamide-adenine dinucleotide phosphate and oxygen. The vasodilatory mechanism of NO can be briefly explained as follows: NO in high concentration is quickly oxidized into toxic nitrite (NO2-) or nitrate(NO3-); however, at low concentrations, NO diffuses into the smooth muscle. Once inside the muscle cells, NO is iron and sulfurfriendly, which enables it to easily combine with and activate the heme of guanylate cyclase, which raises the concentration level of intracellular cGMP. Intracellular cGMP induces vasodilation via many mechanisms. For example, it activates cGMP-dependent protein kinase, which inhibits Ca++ entry into the cell, causes Ca++ to leave the cell, sequestrates Ca++ in the sarcoplasmic reticulum (SR) and reduces intracellular Ca++. In addition, intracellular cGMP activates myosin light chain phosphatase, reduces phosphorylation in the myosin thereby sustaining the vascular smooth muscle tone, and reduces vascular tone [11-14] (Fig. 2, 3). NO combines with oxy-Hb in the blood and to produce met-Hb which is reduced by a reductase and also combines with oxygen and forms NO3- and NO2- which are excreted in urine [15].
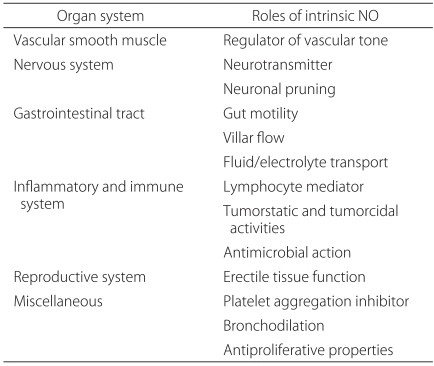
NO works as a biological controller in many organs. One of its most important functions is regulation of the vascular smooth muscle (Table 2). In 1980, Furchgott and Zawadzki first reported that endothelial cell integrity is vital during achetylcholine vasodilation. They were also the first to report the existence of EDRF (endothelial-derived relaxing factor) [16]. In 1987, Palmer et al. demonstrated that the substance produced in normal vascular endothelial cells was in fact NO [17]. Later, they found that chronic inflammatory respiratory patients lacking pulmonary vasodilatory function had reduced endothelial NOS and NO activity [18].
Studies conducted to determine if the administration of NO suppresses vascular smooth muscle constriction and fibroblast proliferation [19] strongly suggest that NO affects vasodilation. NOS is found in three isoforms: neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS). eNOS is primarily known for its role in vasodilation [20,21]. To determine what role NO plays in controlling vascular tone in normal blood vessels, different quantities of the NOS inhibitors, N-monomethyl-L-arginine (L-NMMA), N-nitro-L-arginine methyl ester (L-NAME) or N-nitro-L-arginine (L-NA) were added to different animal lung models using different administration methods. The results showed that when NOS inhibitors were administered, NO production was inhibited and endothelium-derived vasodilation was reduced in healthy blood vessels. Additionally, increased quantities were found to trigger pulmonary vasoconstriction. Furthermore, the effects of L-NAME and L-NA were greater than those of L-NMMA [22-25]. The role of NO in vasoconstriction has also been evaluated through a wide variety of animal test models. More NO production was observed in models in which pulmonary resistance increased due to hypoxia and the administration of angiotensin II than in the normal pulmonary resistance model. In addition, hypoxic pulmonary vasoconstriction was more intense in response to L-NAME and L-NA than normal PVR [23,26,27]. Increased NO reduces pulmonary resistance, but also produces toxic radicals, although less amount produced than in adult respiratory distress syndrome, and NO can lead to blood vessel damage. The balance of cell toxicity and cell integrity is determined by each radical fraction, which determines the final reaction [28-30].
After the discovery of the pathway through which NO affects vascular relaxation and vascular tone, many studies were conducted to identify ways in which NO could be used clinically. Frostel et al. and Pepke-Zaba et al. reported that administration of NO at 40 ppm to adult patients with primary high pulmonary blood pressure could selectively reduce pulmonary resistance without altering the systemic vascular resistance [31,32]. Since then, many clinical studies of various patient groups have shown the utility of NO in the diagnostics, treatment and prognoses of PHT in patients such as newborn babies, as well as those with juvenile hereditary heart disease accompanying PHT, adult respiratory distress syndrome and chronic obstructive pulmonary diseases [3,15,33-39].
Nitric oxide in cardiac surgery
Pulmonary hypertension can occur even after cardiopulmonary bypass because of microembroli, ischemia, sedimentation of blood cells or stress reaction. Existing methods of reducing pulmonary resistance before and after surgery include systemic alkalization, O2 therapy, increased cardiac contractility, stress reaction reduction, anticoagulant drugs and vascular relaxants. The most commonly used drugs are alpha-adrenal receptor antagonists, beta-lactam antibiotics, nitrates, Ca++ channel blockers and prostaglandins. However, these drugs seriously reduce systemic vascular resistance and cause unstable vital signs [40,41]. When PHT cannot be corrected with drugs, extracorporeal circulation is conducted to induce oxygenation; however, there are no special methods of treatment besides symptomatic therapy, and often needed long-term hospitalization.
Following cardiopulmonary bypass, elevated pulmonary resistance does not subside in response to achetylcholine administration, but it does subside in response to NO inhalation. In addition, the administration of NO has been found to increase the cGMP level. Although the vasodilatory effect is not endotheliocyte-dependent, NO has been shown to have a direct relaxant effect [5]. There have also been many reports of NO administration selectively reducing pulmonary resistance during mitral valve replacement due to stenosis, mitral valve replacement accompanied by right ventricular failure and LVAD insertion due to left ventricular failure, heart transplantation and before and after various cardiac surgeries [42-47].
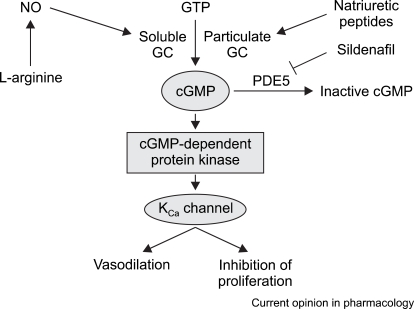
It has also been argued that when compared to prostacyclin, NO is more effective at reducing PHT [44]. However, although the two drugs induce a similar reduction in pulmonary resistance, prostacyclin has a low toxicity and is easier to administer; therefore, it is more commonly recommended [43,46]. When compared to milrinone, NO keeps the heart rate low and better sustains the right ventricular index and systemic arterial pressure [47]. In addition, there was a case report of an allograft liver transplant in which the patient experienced serious PHT mid-operation that was effectively reduced by combined treatment with prostacyclin and NO [48]. In a comparative study of the recently developed inhalant PGI2 drug, iloprost, both NO and PGI2 successfully reduced PHT after a cardiac pulmonary bypass, but it was not determined which drug was most effective. Furthermore, both drugs have similar effects and it has been reported that when used together they have a synergistic effect [49]. Prostaglandin has the same effects as NO, but its mechanism in capillary relaxation is different. Specifically, NO activates cGMP and relaxes the vascular smooth muscles, while iloprost raises the intracellular cAMP level, activates Ca++, opens the K+ channel and reduces pulmonary resistance (Fig. 4). Another advantage of iloprost is that its half-life is longer than that of NO, which results in a low occurrence of rebound PHT during weaning [50].
The use of NO during a perioperative period, especially post-surgery, has produced a variety of responses. Furthermore, the differences in responses have not necessarily been proportional to the dosage amount, but are known to be influenced by many factors related to surgery such as hemodynamic management and ventilation conditions. Accordingly, it is dif ficult to predict the standard effects of NO, which necessitates that drug therapy be selected on a case by case basis to magnify the desired effect and minimize the side effects. Moreover, in the event of intraoperative uncorrectable emergency PHT, NO should be used first, while the use of ECMO should be considered only if NO is not effective [15,39].
Therapeutic use of NO
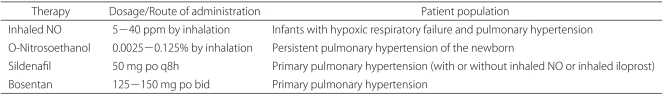
Since it was approved for medical use by the FDA in 1999, NO has been very useful in clinical research. In patients undergoing mechanical ventilation, NO is connected to an artificial ventilation system with deseired flow-rate after regulation of pressure (Table 3). During its administration, NO inhalation and the NO2 levels are monitored just proximal the tracheal tube using an electrochemical analysis machine, as is the expiratory gas flow. Patients without tracheal tubes simply supply NO through an oxygen mask or hood, but expired gas and redundant gas must be carefully removed using a vacuum [51].
Although treatment with NO at levels below 0.8 ppm has been reported to have pulmonary vasodilatory effects [52], treatment with 2-80 ppm is usually most effective [53,54]. As a result, treatment generally starts at 5-20 ppm, and the dosage is then increased to a maximum of 80 ppm if there is no reaction within a few minutes. However, it is recommended that less than 40 ppm be administered during chronic NO treatment [7,51]. Overall, it is important to administer the lowest effective dose while continuously monitoring the NO2 level and maintaining the inhaled oxygen level between 0.4 and 0.6 to minimize oxygen and NO2 toxicity [55].
During NO treatment, it is recommended that the central venous pressure, pulmonary arterial pressure, systemic arterial pressure and systemic oxygen saturation be measured. However, if this is not practical, it is essential to monitor the systemic arterial pressure and oxygen saturation. Furthermore, post-operative patients or ICU patients need to be provided with sedatives as well as additional hyperventilation.
At 24 hours after the start of treatment, the NO can be reduced and weaning may be considered depending on the patient's condition. At this point, the NO concentration can be slowly reduced from 10 ppm to 0-5 ppm. During this initial weaning period, the pulmonary arterial pressure and systemic arterial pressure should be carefully monitored. An increase in pulmonary arterial pressure and a decrease in systemic arterial pressure within the first 1-2 min signifies a failure in weaning, indicating that NO administration must not be stopped. Additionally, if the patient condition is unstable during weaning, re-administration of NO at the previous level should be considered. After re-administration, weaning may be considered after 24 hours. NO weaning should be conducted progressively so that the endotheliocytes can recover from the endogenous NO inhibition and down-regulation from the exterior NO administration [56]. Indeed, sudden weaning can poses the risk of causing serious hypoxia and rebound PHT. When weaning from NO administration, temporary pulmonary arterial pressure or vascular resistance may rebound and increase, but this is different from weaning-failure. Rather, this happens several minutes after stopping NO administration. Although the pulmonary arterial pressure increases, the systemic arterial pressure and the oxygen saturation often remain the same, and the pulmonary arterial pressure then decreases after several minutes. It is believed that this phenomenon occurs due to a decrease in the production of endogenous NO.
The length of the administration period cannot be set uniformly, but must be set differently for each patient. For an adult, NO is generally administered for several days to several weeks, but the average administration period is five days [57]. When the pulmonary resistance is greater than six wood units, NO should be administered for more than 72 hours while taking care when decreasing the right ventricle afterload [51]. Patients with post-surgery PHT that is unresponsive to NO treatment do not have a good prognosis. Additionally, juvenile patients with post-surgery PHT often experience cyanosis. If such patients are unresponsive to NO, it is important to check for pulmonary venous occlusion and any pulmonary arterial malformations [58]. Through the preoperative administration of NO in patients preparing for a heart transplant, we can check the responsiveness of the pulmonary vasculature to determine if surgery is possible and to evaluate the prognosis [59].
Recently, there has been increased research into treatment with liquid NO dissolved into normal saline and injected into the brachial artery. The results of these studies have shown that the administration of 0.75-6 µmol NO causes rapid radial artery relaxation and stabilizes the decrease in resistance for 20 seconds. Additionally, these studies have shown that large quantities of plasma NO2 and NO3 were produced in response to this treatment, as well as s-nitrosothiol (RSNOs), which is assumed to be the substance that prolongs pulmonary vasodilation [60].
Toxicity of nitric oxide
Before NO was known for its utility, it was known as an environmental toxin with great cytotoxicity. For example, cigarette fumes are known to contain an average of 400 ppm of NO and polluted air often contains up to 0.2 ppm of NO2. Endogenous NO is also believed to be the cause of oxidation-mediated tissue damage in septic shock, reperfusion injury and pulmonary inflammatory reaction [61]. NO can be toxic because gaseous NO reacts with O2 to create NO2, which can cause lung damage in levels proportional to the concentrations of oxygen and NO. When there are high concentrations of oxygen and NO, large quantities of NO2 are produced; therefore, if the level of NO cannot be reduced it is necessary to reduce the level of oxygen to prevent toxicity. In general, more than 5 ppm of NO2 can cause pulmonary toxicity [62]. In animals, NO2 has been reported to cause damage in Hulle cells, epileptic contractions and fibrostic lesions [63,64]. Additionally, although it is known that chronic exposure to NO can lead to respiratory tract hypersensitivity, bronchitis and cancer, there have not been many reports of pulmonary toxicity in response to short-term exposure [63].
As mentioned above, inhaled NO is rapidly absorbed into the blood stream, where it combines with the heme in Hb and oxides to create Met-Hb. This oxidated Hb has less ability to deliver oxygen. Accordingly, many clinical studies have been conducted to evaluate the effects of this increased met-Hb concentration, but there have been no clinical reports of seriously reduced function of oxygen delivery. Rather, in patients administered high levels of NO, an increase in met-Hb levels is generally observed, but these levels drop immediately after the NO level is reduced [11,12,15,51,54,65].
Liquid NO reacts to oxygen much faster than gaseous NO to form the strong oxidant, NO3-, which can cause tissue damage via mediated oxidation [66-68]. As previously mentioned, when NO is used to treat adult respiratory failure and sepsis, it is important to realize that the cGMP-mediated vascular smooth muscle vasodilation and toxic radical production can actually worsen any inflammatory reaction, which can result in sepsis developing into septic shock.
The mechanism by which the effects of NO occur has not yet been fully elucidated; however, it is known to induce toxicity in platelets [69]. Additionally, it is dangerous to use NO in premature infants and child patients with intraventricular hemorrhage because NO has atrophic, nonproliferative properties that cause platelet inhibition [69]. When NO is used for the treatment of such cases, it is important to be mindful of NO2- production, the NO and oxygen levels, systemic inflammation and hemorrhagic tendencies. Specifically, the level of NO2- must be kept below 5 ppm. Additionally, the level of oxygen must be kept low while maintaining a high flow rate. Furthermore, the soda lime filter must be checked, the surrounding air must not be polluted by NO2- and the patient's NO2- level and met-Hb level must be monitored continuously.
Other therapeutic modalities for pulmonary hypertension
O-nitrosoethanol (ENO)
A portion of NO is transformed into RSNO, which is resistant to oxidation and does not become inactive. As a result, RSNO can react continuously. ENO does not react with oxygen or NO; therefore, its presence leads to a notable increase in endogenous RSNO in the alveolus and the respiratory tract. Accordingly, ENO inhalation can immediately reduce hypoxic pulmonary vasoconstriction, and there is no increase in rebound pulmonary pressure or decrease in oxidation when weaning from ENO. Moreover, cardiac output is stabilized, reduced systemic blood pressure is infrequent and the ventilation/perfusion ratio imbalance is mitigated, therefore, ENO is effective for the treatment of PHT accompanied by hypoxemia or accompanied by right ventricular failure [70].
Pulsating ENO inhalation is reported effective at treating PHT and oxidation because this treatment maximizes the effect of sending gas in well-ventilated bronchopulmonary segments and helps NO be exhaled during the rebreathing ventilation cycle [71].
Sildenafil
Sildenafil is an oral Phosphodiesterase (PDEs)-5 inhibitor that is known to be a safe and effective substitute for NO [72-74]. PDE inhibitors prevent the destruction of intraceullar cGMP, which leads to heightened pulmonary vasodilation [74] (Fig. 4). The use of sildenafil alone or with NO increases the cardiac index, brings no change to the systemic blood pressure, and is effective when administered as a one-time oral medication. Other inhibitors such as tadalafil and vardenafil also act as pulmonary vasodilators, but do not increase the effects of oxidization [75]. Recent long-term studies have reported that there is a strong synergistic effect when tadalafil and vardenafil are administered together with iloprost [76].
Bosentan
The endothelin (ET) receptor-inhibitor, bosentan, also induces hemodynamic and functional improvements in PHT patients [77]. The effects of bosentan occur via the blockage of ET receptors and increased internal activity of endogenous NO. The effects of bosentan can be magnified when it is administered in combination with NO [78].
Natriuretic peptides
Similar to NO, atrial natriuretic peptides (ANP) and brain natriuretic peptides (BNP) elevate intracellular cGMP, which in turn controls the vessel tone and affects vascular relaxation. ANP is produced in the atrium whereas BPN is produced in the ventricle. Both ANP and BNP have half lives and can only elevate cGMP to a certain point. Indeed, if the BNP level rises above the necessary level, it signifies a bad prognosis [79]. However, during acute hypoxia, ANP and BNP production increases, which reduces pulmonary resistance and the pressure load in the right ventricle, thereby preventing right ventricular failure. Even during chronic hypoxia, the blood concentration of ANP and BNP is high. Extrinsic ANP also reduces acute hypoxic pulmonary vasoconstriction and right-ventricular strain, while BNP shows similar effects [80].
BNP is used for diagnostic purposes when there are elevated plasma concentrations, left ventricular dysfunction, severe worsening of the right ventricular dysfunction and PHT accompanied by chronic pulmonary disorders [81-83]. Additionally, BNP is used to treat left ventricular dysfunction patients by reducing pulmonary resistance. In mitral disorders, BNP attenuates PHT. In addition, it has been reported that the administration of BNP with sildenafil reduces pulmonary resistance; however, more research on that topic is needed [84].
Overall, NO inhalation has been shown to effectively reduce pre- and post surgical PHT. Additionally, NO can be used during liver-transplants and ventilation therapy for chronic pulmonary disorder patients as well as in cardiac patients. However, NO inhalation is required a complex delivery device an daccurate toxicity-monitoring device. Additionally, NO2- and NO3- formed during treatment can have toxic side-effects and the chronic administration of NO has another problem. To overcome these limitations, the use of sildenafil and other PDE inhibitors, as well as ANP and other orally/intravenously administered drugs is being evaluated. Future studies should enable the development on new drugs and liquid forms of NO that can overcome the side-effects and systemic hemodynamics.
 XML Download
XML Download