

 PDF
PDF Citation
Citation Print
Print


Introduction
Survivin is located in chromosome 17q25, and survivin is a new member of the inhibitor of apoptosis (IAP) family. It is predominantly expressed in fetal tissue, but it is also found in many common human cancers [1-4]. Survivin inhibits the terminal effectors of apoptosis and it also inhibits the induction of apoptosis by Fas, Bax and caspases [1-4].
In addition, survivin has emerged as a unique regulator of cell death through its response to growth factors, such as mitogen-activated protein kinase (MAPK) [1-4]. The transcriptional c-Myc is an oncogene that lies downstream of the MAPK pathway, suggesting a possible role for transcriptional c-Myc in survivin's regulation [2,5-7]. The MEK/ERK pathway can also modulate the activity of many proteins involved in apoptosis such as survivin [1-6]. p53 couples apoptosis to mitogenic signals and the survivin pathways [3-7]. Survivin contains a canonical CpG island that has been described as being epigenetically regulated. Recently there was a report showing that demethylation of the survivin promoter by decitabine results in p53-dependent survivin repression and that p53 binding can be inhibited by DNA methylation. De-repression by methylation is a general mechanism of p53 regulation. Demethylation induced by decitabine is traditionally thought to be active in tumors by allowing the re-expression of tumor suppressor genes [8]. Wang and Dick suggested that oncogenic mutation and epigenetic defects could be inherited from transformed stem cells, giving rise to some populations that have been observed in many malignant tumors [9]. As it is speculated that stem cells can possess neoplastic-like properties and the expressions of the survivin-apoptotic pathway may show the same properties in stem cells and cancer cells [9-11], this study attempted to elucidate the signal transduction pathway of stem cells by a demethylating agent, 5-aza-2'-deoxycytidine (ADC), to analyze the survivin, MEK/ERK, c-Myc and p53 gene expressions.
Go to : 
Materials and Methods
Chemicals
Dulbecco's modified Eagle's medium (DMEM), 0.25% trypsin-EDTA solution, sodium bicarbonate solution, bovine serum albumin (BSA), 3-[4,5-dimethythiazol-2-yl]2,5-diphenyl tetrazolium bromide (MTT), propidium iodide, ethidium bromide, acridine orange, rhodamine 123, agarose, β-actin antibody and ADC were purchased from Sigma Chemicals Co. (St. Louis, MO, USA). Fetal bovine serum (FBS) and antibiotic/antimycotic solution were purchased from Gibco (Gibco, USA). Sodium phosphate (monobasic and dibasic), sodium chloride, sodium hydroxide, sodium carbonate, hydrochloric acid and methanol were purchased from Sisco Research Laboratories (Mumbai, India). The primary antibodies against c-Myc, p53 and survivin were purchased from Dako (Amsterdam, Netherlands), ERK and MEK were purchased from Genzyme (San Francisco, CA, USA).
Human adipose-derived mesenchymal stem cells
Adipose-derived mesenchymal stem cells (ASCs) (from Dr. Young-il Yang) were isolated from the lipoaspirates of patients undergoing cosmetic liposuction, as described by Zuk et al. [12]. In accordance with a consensus reached by the investigators, the author referred to this adherent cell population as ASCs. Briefly, the tissue was digested for 45 min with 0.075% collagenase I (Gibco, USA). The stromal-vascular fraction was separated from the remaining fibrous material and the floating adipocytes by centrifugation at 300 g. The sedimented cells were filtered through a 100 µm pore filter. Erythrocyte contamination was reduced by performing density gradient centrifugation with Bicoll (Biochrom). High contamination with erythrocytes was found to markedly decrease cell adherence and proliferation. A preceding density gradient separation procedure provided a better yield of adherent cells than did treatment with an erythrocyte lysing buffer. For initial cell culture and expansion of the cells, DMEM with a physiologic glucose concentration (100 mg/dl) supplemented with 10% fetal calf serum (FCS; PAA) was used. The primary cell isolates and cultured cells were characterized as described by Brzoska et al. [13]. The cultured ASCs were CD29+, CD44+, CD49a+, CD73+, CD90+, CD105+, CD166+, and CD14-, CD31-, CD45-. The in vitro differentiation potential of the ASCs was proven by using specific media. For these experiments, the 2nd-5th passage of the ASCs was used.
Cell culture
The ASC line was from Dr. Young-il Yang. The cells were grown in T75 culture flasks containing DMEM supplemented with 10% FBS, 100 units/ml penicillin and 100 units/ml streptomycin in a humidified 5% CO2 atmosphere at 37℃. Upon reaching confluence, the cells were detached using Trypsin-EDTA solution.
Cell proliferation assay
Proliferation of the ASCs was assessed by MTT assay. The cells were plated in 24-well plates at a concentration of 5 × 104 cells/well. Twenty-four hr after plating, they were washed twice with 500 µl of serum-free medium and then they were starved by incubation in serum-free medium for an hour at 37℃. After starvation, the cells were treated with ADC at 0.5, 1, 2, and 2.5 um concentrations for 24, 48, and 72 hr. At the end of treatment, the media from the control and ADC-treated cells were discarded and 500 µl of MTT containing DMEM (0.5 mg/ml) was added to each well. The cells were then incubated for 4 hr at 37℃ in a CO2 incubator. The MTT-containing medium was then discarded and the cells were washed with 1× phosphate-buffered saline (PBS; 1 ml). The crystals were then dissolved by adding 500 µl of solubilization solution and this was effectively mixed by pipetting up and down. The spectrophotometrical absorbance of the purple blue formazan dye was measured using a microplate reader at 620 nm. The optical density of each sample was then compared with the control optical density and the graphs were plotted. Based on MTT assay, the dose of 1 um ADC treatment was selected for further studies because this dose was below the IC50 (50% inhibitory concentration) value of ADC for the ASCs.
ADC treatment
The cells were subjected to cell cytotoxic assay against ADC with various dosages of ADC. A 1 uM dosage was found to be most appropriate dosage for this study with using ASC. The appropriate number of the cells (1 × 104) was seeded onto 96-well plates and the cells were treated with 1 uM dose of ADC for 24, 48 and 72 hr. Afterwards the cells were detached with trypsin-EDTA and the plates were rinsed and the cells were collected. The cells were stained with 0.4% trypan blue. The viable cells were calculated under the hemocytometer.
RNA isolation
The ASC cells from the same isolation and in the same passage were identically treated. The cells were grown to subconfluence (70%), washed and kept in serum-starved for 2 hr. In brief, the cells were lysed using 10 mM Tris pH 7.4, 0.1% SDS, 0.1% Tween 20, 0.5% TritonX100, 150 mM NaCl, 10 mM EDTA, 1 M urea, 10 mM NEM, 4 mM benzamidine, and 1 mM PMSF and then they were collected by scraping.
The total RNA was isolated from each of the samples after lysis in quanidinium isothiocyanate and phenol extraction using the commercial kit (Trizol, Invitrogen Laboratories, SanDiego, USA).
Reverse-transcription polymerase chain reaction (RT-PCR) for detection of survivin mRNA
The cDNA was synthesized from 4 µg of total RNA in a 25 µl reaction mixture containing 6 µl of 5X reverse transcriptase reaction buffer, oligo (dT) (100 pmol/ul) 1µl, 10 mM dNTP 4 µl, 40 unit/µl of RNAsin and 0.5 µl (200 units/L) of Moloney leukemia virus reverse transcriptase (MMLV RTase). The mixture was incubated at 4℃ for 60 min heated to 94℃ for 3 min and then chilled on ice. To test the cDNA integrity, GADPH was amplified in each of the samples. All the oligonucleotide primers were synthesized by and purchased from the Bioneer Co (Taejun, Korea). The oligonucleotide primers were solubilized in TE buffer to the concentration of 100 pmol/µl. The aliquots were diluted to 10 pmol/µl before use. To ensure that the RNA was not degraded, a PCR assay with primers specific for beta-actin cDNA was carried out for each of the cases under the following cycling conditions: 25 cycles of 30 sec at 94℃, 30 sec at 60℃ and 30 sec at 72℃ with pre-denaturation for 2 min at 94℃ and post-extension at 72℃. A 10 µl, aliquot of each reaction mixture was fractionated on 1% agarose gel and the results were visualized with ethiumbromide staining. cDNA was synthesized by random priming using less than 1 µg of total RNA as a template. As an internal standard, a fragment of human G3PDH was amplified by PCR using the forward primer 5'-ACC ACA GTC CAT GCC ATC AC-3' and the reverse primer 5'-TCC ACC ACC CTG TTG CTG TA-3' which gave a PCR product of 450 bp. The PCR amplification was performed in a final volume of 20 µl which was comprised of 1 µl of the reverse transcriptase reaction mix, 3 pmol each of the 5' and 3' primers for the survivin or the G3PDH genes, 160 µM of deoxy nucleotide triphosphate, 1 mM MgSO4 and 0.3 units of Taq DNA polymerase in reaction buffer. The PCR was performed with an initial denaturation step at 94℃ for 2 min followed by 30 cycles of 20 sec at 94℃ for denaturation, 30 sec at 68℃ for annealing and 45 sec at 72℃ for extension. The final extension step was prolonged to 5 min at 72℃. The PCR products were separated on 2% agarose gels by electrophoresis.
Direct DNA Sequencing of the PCR products
The wizard plus SV miniprep kit (Quazen, Munik, Germany) was used for the template DNA preparation for sequencing after subcloning the RT-PCR products. An automatic DNA sequencer (ABI sequencer 3700, Macrozen, Seoul, Korea) was used for sequencing. The sequence data were analyzed by the NCBI (NIH, USA) Blast search program.
Western blot analysis
The western blot analysis for the protein expression in the ASC cells was assessed using the following method. Approximately 50 µg of protein was mixed with an equal volume of 2× sample buffer, this was boiled for 5 min at 95℃, cooled, loaded on each lane of an 8-15% polyacrylamide gel, and separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) at room temperature. The resolved proteins were electrophoretically transferred to nitrocellulose membranes which were then blocked in 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T) for 1 hr at room temperature, and then the proteins were probed with the following primary antibodies c-Myc (dilution 1 : 500), p53 (rabbit polyclonal antibody at a dilution of 1 : 1,000), ERK (rabbit polyclonal antibody at a dilution of 1 : 500), p53 (rabbit polyclonal antibody at a dilution of 1 : 1,000), MEK (mouse monoclonal antibody at a dilution of 1 : 500), survivin (mouse monoclonal antibody at a dilution of 1 : 500), and β-actin (mouse monoclonal antibody at a dilution of 1 : 2,000), this was performed overnight at 4℃. Blots were then extensively washed with TBS-T and they were incubated with the respective (anti-goat, anti-rabbit and anti-mouse) horseradish peroxidase-labeled secondary antibodies (Genei, Bangalore, India) at dilution of 1 : 2,000 for 1 hr at room temperature. After thorough washes in TBS-T, the bands were visualized by treating the membranes with 3,3'-diaminobenzidine tetrahydrochloride (Sisco Research Laboratories). The membranes were photographed and next quantified using image analysis software (NIH, Bethesda, MD, USA). The densitometry data presented in the bar graphs are the 'fold change' as compared with that of the control in each case.
Statistical analysis
The results were analyzed using SPSS software (version 17, Stanford, SC, USA) and the difference between the protein levels of the treated and untreated groups was evaluated using Student's t-test. Multiple groups were compared using the oneway analysis of variance (ANOVA) test. P values < 0.05 were deemed statistically significant.
Go to :
Results
Survivin mRNA
The ASCs expressed survivin mRNA before ADC treatment and they still expressed survivin mRNA after treatment, and the level was decreased slightly after 48 hr and it was restored after 72 hr (Fig. 1).

The protein expression of the genes
The immunoblotting showed increased levels of the protein expression of survivin up to 48 hr after treatment. However, its level after 72 hr was decreased to lower than that before treatment. The levels of p-MEK and p-ERK were increased time-dependently, however these levels at 72 hr were decreased to lower than that of before treatment. The expression of unphosphorylated ERK was not changed. The expression of c-Myc was elevated after ADC treatment and it was highest at 48 hr after treatment. The expression c-Myc at 72 hr was higher than that before treatment (Fig. 2). The p53 expression was elevated time-dependently up to 72 hr. ADC treatment significantly increased the expression of MEK, ERK, c-Myc, p53 and survivin time-dependently as was evident from the immunoblotting and the corresponding densitometry data (P < 0.05). The survivin expression was significantly correlated with those of c-Myc (P = 0.003) and p-ERK (P = 0.001) (Fig. 3).
 | Fig. 2The expression of proteins on ASC with ADC treatment by Western blotting. All proteins are expressed before treatment, and show various change of their expressions after treatment.
|
 | Fig. 3Protein expression analysis of p-MEK, p-ERK, c-Myc, p53, and survivin in control and ADC-treated ASC as assess by immunoblotting and by their respective densitometry data. The comparison of each protein level at 24, 48, 72 hr after and before ADC treatment is as follows: P-p-MEK = 0.08, P-p-ERK = 0.001, P-c-Myc = 0.003, P-p53 = 0.004, P-survivin = 0.031, P-p-MEK-survivin = 0.075, P-p-ERK-survivin = 0.010, P-c-Myc-survivin = 0.003, P-p53-survivin = 0.117.
|
Cell morphology
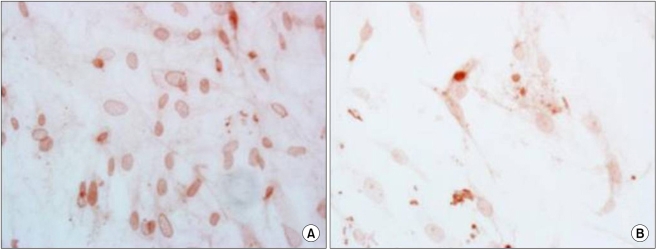
ADC inhibited the population growth of the ASCs and the percentage of apoptotic cells was increased at 24, 48, and 72 hr after treatment (Fig. 4). The immunohistochemistry showed that the nuclear expressions of ERK and survivin were increased after ADC treatment (Fig. 5, 6).
 | Fig. 4The morphology of ASC. Before ADC treatment (A), 24 hr after ADC treatment (B), 48 hr after ADC treatment (C), 72 hr after ADC treatment (D): ADC inhibits population growth of the ASC and the percentage of apoptotic cells increases 24, 48, and 72 hr after treatment.
|
Go to :
Discussion
The role of survivin associated with the MAPK pathway, c-Myc, and p53 has been reported for malignant tumor, but it still is not fully understood [1-6]. Recently, the concept of "cancer stem cell" has emerged on the basis that some populations of cancer cells have stem cell-like properties [14-17]. The aim of this study was to investigate the effect of the demethylating agent ADC on survivin and its related proteins in a stem cell pathway using ASCs. In the current study, the ASCs had expressed survivin mRNA before ADC treatment and they still expressed survivin mRNA after treatment, and the level of survivin mRNA was decreased slightly after 48 hr and it was restored after 72 hr. This finding is consistent with Fukuda and Pelus' report [18]. They reported that survivin is highly expressed during embryonic development, but it is negligibly expressed in most adult tissues. The idea Fukuda has put forth is that survivin may contribute to stem cell homeostasis. Accordingly, earlier studies suggested that survivin was required to preserve the viability of hematopoietic progenitor cells [19].
DNA methylation is a reversible process in which genes can be demethylated and restored to their original expression and function [20-22]. The DNA methylating agent, ADC is thought to inhibit the enzymes that methylate the cytidine residues in DNA and demethylate the methylated DNA [21,22]. This study showed a gradually increased level of the protein expression of survivin up to 48 hr after treatment. However, its level at 72 hr was lower than that before treatment. This study disclosed the apoptosis was increased maximally at 72 hr after ADC treatment, while the nuclear expression of survivin was increased. This result can explain the translocation and subcellular localization of survivin, that is, when survivin is in the cytoplasm it acts to suppress apoptosis, but when survivin is in the nucleus it acts as a chromosomal passenger protein at the G2/M checkpoint [1-4,11,15]
In this study, the p53 expression was elevated time-dependently up to 72 hr after treatment. This finding is consistent with the morphological change of increasing apoptosis. p53 protein inhibits the growth of tumors by arresting cell proliferation and inducing apoptosis [14,15]. The current study showed the survivin expression was increased up to 48 hr after treatment together with a gradual increase of p53. However, Nabilsi et al. reported that demethylation of the survivin promoter by decitabine resulted in p53-dependent survivin repression and that p53 binding can be inhibited by DNA methylation in the endometrial cancer cell line [8]. This opposite result might suggest that the expression of survivin in ASCs was regulated via a different signal pathway from that of the endometrial cancer cell line. There was a report that most methylated ovarian cancer is survivin-negative, which means demethylation can make some cancers express survivin [23].
The levels of phosphorylated-MEK and -ERK were increased time-dependently, however the levels 72 hr after ADC treatment were decreased to lower than that before treatment. c-Myc was elevated after ADC treatment and its highest level was noted 48 hr after ADC treatment. The expressed c-Myc level at 72 hr was higher than that before treatment.
In this study, ADC treatment significantly increased (P < 0.05) the expressions of MEK, ERK, c-Myc, p53 and survivin, as is evident from the immunoblotting and the corresponding densitometry data. Especially the expression of survivin was significantly correlated with those of c-Myc (P = 0.028) and ERK (P = 0.010). There are a few previous reports that support the results of this current study, although most of the previous reports were studied cancer. Carter et al. [24] reported that the survivin expression in leukemic cells is regulated by cytokines and differentiation-inducing agents and that the modulation of major signal transduction pathways, such as the MEK/ERK at the mRNA and protein levels. Arango et al. [7] described that the c-Myc expression increases the cellular sensitivity to DNA damage-induced apoptosis. Seoane et al. [10] presented that the c-Myc expression is also important in determining the action of p53 in response to DNA damage. A high level of c-Myc can favor the apoptosis response to p53 [9]. Accordingly, this study showed the p53, and c-Myc related apoptosis in ASCs that were damaged by ADC treatment, yet there was the concomitantly upregulation of the anti-apoptotic protein, survivin. This controversy can be explained by the reports of Yuan and Filion that the upregulation of survivin occurred during the immotalization of human adult cells [25,26]. The ASCs utilized in this study might have immortalized properties, although precise further evaluation will be needed to verify the current results. Besides, it should be considered that the survivin-pathway might be not universal but rather, it is cell or tissue-specific and demethylation is also a mechanism affected by time and the internal or external environments.
In conclusion, the overexpression of p-MEK and ERK, p53, and c-Myc increased the survivin protein expression of the demethylated ASCs. These results suggest that demethylation could alter the survivin expression and p53, c-Myc and the MAPK pathway might play a role in survivin's regulation in adult mesenchymal stem cells. Therefore, survivin helps maintain the homeostasis of adipocyte-derived stem cell. This study presents evidence that the expression of multiple gene pathways controls this response, including the MEK, ERK, c-Myc and p53 genes. Further study with stem cells will be needed to verify these results.
Go to :
 XML Download
XML Download