

 PDF
PDF Citation
Citation Print
Print


Introduction
Propofol has a myocardial protective effect through a range of mechanisms, such as oxygen free radical scavenging [1-3], blocking of the calcium channel by inhibiting the L-type calcium channel [4,5], and inhibiting neutrophil activation [6,7]. The myocardial protective effect of propofol has been identified by a variety of methods using an ischemia/reperfusion (I/R) injury model [1-3,7,8]. However, some technical problems make it difficult to perform a long-term follow-up of the myocardial protective effect. Some reports have shown that myocardial infarct size and neutrophil adherence peak at 24 and 48 h, respectively, after an I/R injury [9,10]. During the late reperfusion period, myocardial apoptotic cell death develops progressively until 72 h [10]. Therefore, long-term observations will be needed to determine whether myocardial protection is a temporary or sustained effect [11-13].
In this study, a regional myocardial I/R injury in an in vivo rat heart model was used to determine the effects of a clinically relevant propofol concentration during the periischemic period of anesthesia. After 48 h of reperfusion, the hemodynamic function, infarct size, and cardiac troponin-I (cTn-I) level were checked to determine whether propofol had a delayed myocardial protective effect against a regional myocardial I/R injury in an in vivo rat heart model.
Materials and Methods
Animal preparation
Male Sprague-Dawley rats weighing between 220 and 240 g were used. All animals were maintained in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health in 1996.
The animals received general anesthesia with an intramuscular injection of 50 mg/kg of ketamine (Ketamine Hcl®, Huons, Seoul, South Korea) and 3 mg/kg of zylazine (Rompun®, Bayer, Seoul, South Korea). The rats were judged to be awake from the anesthesia once their tails moved during the procedure, and then 25 mg/kg of ketamine and 1.5 mg/kg of zylazine were injected to maintain the level of anesthesia. After anesthesia was induced, a heparin-treated 24 G catheter was inserted into the femoral vein, and each drug was infused according to the protocol. The trachea was intubated, and the rats were ventilated mechanically (tidal volume: 8 ml/kg, respiratory rate: 80 strokes/min) with ambient air using a volume-controlled rodent ventilator (Harvard Rodent Ventilator Model 607, Harvard, Holliston, MA, USA). The arterial blood pH and gases were maintained within normal physiological limits (pH: 7.3-7.4, PaCO2: 30-40 mmHg, PaO2: 60-100 mmHg).
A paramedian sternotomy was performed, and the pericardium was opened to expose the heart. For the I/R experiments, a 4-0 black silk, nonabsorbable suture was passed around the left coronary artery (LCA) territory to induce regional myocardial ischemia. The coronary artery was occluded by pulling a snare through a small vinyl chloride tube and clamping it with a mosquito hemostat. Reperfusion was achieved by releasing the clamp. Myocardial ischemia was confirmed by the appearance of regional epicardial cyanosis over the myocardial surface and by arrhythmia. After 25 min of ischemia, the myocardium was reperfused by loosening the snare, which was maintained for 48 h of reperfusion. Successful reperfusion was confirmed by the disappearance of epicardial cyanosis and the production of epicardial hyperemia and arrhythmia after releasing the snare. The body temperature was monitored with a rectal thermometer and maintained at 36-38℃ using a heating pad.
Experimental protocols
Sixty-four rats were divided randomly into four groups. The sham group received a bolus infusion of 2.5 mg/kg of profopol (Fresofol® 2%, Fresenius Kabi, Homburg, Germany) followed by a continuous propofol infusion at a rate of 200 µg/kg/min, which was maintained until 30 min after the sham operation. The LCA was not occluded. The control group received 0.3 ml of normal saline intravenously followed by a continuous infusion of normal saline at a rate of 1.5 ml/h. The propofol group received a bolus infusion of 2.5 mg/kg of profopol followed by a continuous propofol infusion at a rate of 200 µg/kg/min. The intralipid group received a bolus infusion of 0.3 ml of 2% intralipid (20% Intralipid®, Fresenius Kabi, Sweden) followed by a continuous infusion of 2% intralipid at a rate of 1.5 ml/h. A 25 min LCA occlusion was produced followed by 48 h of reperfusion. Each drug was administered intravenously 5 min before the ischemic insult, and continuous drug infusion was maintained until 5 min after reperfusion.
An additional experiment was performed when a rat died during an experiment or during the postexperimental care period. An additional experiment was required for five, zero, six, and four rats in the control, sham, propofol, and intralipid groups, respectively. Two and four rats in the control and intralipid groups, respectively, required additional anesthesia. The hemodynamic function and infarct weight were checked in 10 rats of each group. The cardiac troponin-1 (cTn-I) level was also checked in six rats from each group.
Measuring hemodynamic variables and myocardial contractility
After the 48 h reperfusion, the rats were anesthetized with 25 mg/kg of ketamine and 1.5 mg/kg of xylazine. The right common carotid artery was cannulated with a 2-Fr Millar catheter (Model SPR-407, Millar Instruments Inc., Houston, TX, USA) and advanced into the left ventricle (LV) to measure heart rate (HR), left ventricular systolic pressure (LVSP), left ventricular end diastolic pressure (LVEDP), the peak rate of intraventricular pressure increase (+dP/dtmax), and the peak rate of intraventricular pressure decrease (-dP/dtmax). The systolic and diastolic pressures were checked after repositioning the catheter to the ascending aorta.
Determining the areas at risk and infarct size
After analyzing hemodynamic function, the LCA was occluded again, and 1 ml of 2% Evans blue dye was injected into the pulmonary artery to determine the areas at risk (AAR). The perfused myocardium was stained with Evans blue dye, whereas the occluded vascular bed was not. The atrium, right ventricle, and major vessels were removed from the heart, and the AAR was separated from the non-ischemic area. The AAR was cut into small pieces and incubated in 0.5% 2,3,5-triphenyltetrazolium chloride (TTC) at 37℃ for 15 min. The non-infarcted myocardium was stained a deep red color with TTC, and the infarcted area (IA) remained a pale yellow. The sections were fixed for 24 h in 2% formalin, and the LV, AAR, and IA weights were determined. The AAR and IA were expressed as a percentage of the LV and AAR weights, respectively.
Measuring cTn-I
Twenty-four rats were assigned to the four groups to measure cTn-I levels. The thoracic cavity was opened to expose the heart and collect a 2 ml blood sample from the right ventricle. The serum was collected and stored at -75℃ until analysis. The serum cTn-I level was determined using an enzyme linked immunosorbent assay kit from Life Diagnostics (cat. no. 2010-2-HS, West Chester, PA, USA). The cTn-I activity was detected by spectrophotometry (Infinite® F200, Tecan Group Ltd., Mönnedorf, Switzerland) at 460 nm after reacting the serum sample on a cTn-I antibody-coated plate.
Statistical analysis
All values are expressed as the mean ± SD. All statistical analyses were performed using SPSS software (ver. 12.0 for Windows, SPSS Inc., Chicago, IL, USA). The differences between groups were evaluated using a Friedman one-way analysis of variance and a post hoc Tukey's multiple comparison test. Statistical significance was defined as P < 0.05.
Results
Hemodynamic function following I/R injury
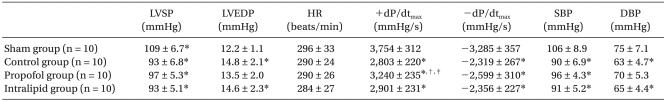
The hemodynamic measurements recorded after 48 h of reperfusion revealed LV dysfunction, as demonstrated by the increased LVEDP (P = 0.01) and decreased LVSP (P = 0.01), +dP/dtmax (P = 0.0001), and -dP/dtmax (P = 0.0001) in the control group compared to the sham group. Propofol significantly inhibited the decrease in +dP/dtmax compared to the control group (P = 0.002) and intralipid group (P = 0.01) (Table 1).
Myocardial infarct size
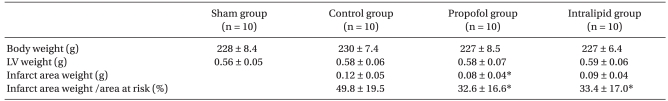
No significant differences between the three groups were observed in the AAR/LV induced by occluding the LCA territory. The IA weights in the control, propofol, and intralipid groups were 0.12 g, 0.08 g (P = 0.031 vs. control group), and 0.09 g, respectively. The IA/AAR in the control, propofol, and intralipid groups was 49.8%, 32.6% (P = 0.014 vs. control group), and 33.4% (P = 0.045 vs. control group), respectively. No significant differences in IA weight and IA/AAR were found between the propofol and intralipid groups (Table 2).
Serum cTn-I levels
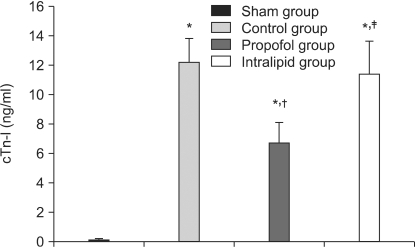
The serum cTn-I levels in the sham, control, propofol, and intralipid groups were 0.087 ng/ml, 12.2 ng/ml (P = 0.0001 vs. sham group), 6.7 ng/ml (P = 0.0001 vs. sham group, P = 0.0001 vs. control group), and 10.4 ng/ml (P = 0.0001 vs. sham group, P = 0.005 vs. intralipid group), respectively (Fig. 1). The I/R-induced serum cTn-I level was reduced by propofol infusion during the peri-ischemic period.
Discussion
Propofol improved myocardial contractility, reduced myocardial infarct size, and decreased serum cTn-I levels after a regional I/R injury in an in vivo rat heart model after 48 h of reperfusion. It has been reported that necrotic cell death reaches a peak at 24 h, whereas apoptotic cell death in the peri-necrotic myocardium develops progressively to 72 h during the late reperfusion period [10]. Although many reports have addressed the myocardial protective effect of propofol, few reports have examined the long-term effects. Myocardial infarction was extended progressively; therefore, the hemodynamic parameters, infarct size, and cTn-I were measured after 48 h of reperfusion to identify the delayed myocardial protective effect of propofol. Propofol improved cardiac function and reduced myocardial infarct size following a regional I/R injury in an in vivo rat heart model after a 48-h reperfusion.
Of the hemodynamic parameters, +dP/dtmax represents myocardial contractility, and +dP/dtmax improved significantly in the propofol group compared to the control group, indicating that the myocardial protective effect of propofol was maintained after a 48 h reperfusion. The effect of myocardial protection was not related to intralipids. Diastolic function is another important myocardial contractility factor, which is represented by -dP/dtmax. However, in this study, -dP/dtmax did not improve significantly in the propofol group compared to the control group.
Although quantitating myocardial infarct size is subjective, it is a useful means to evaluate myocardial infarct size. A prolonged reperfusion time of 24-72 h allows for a more precise histological quantitation of necrosis than do shorter times and excludes the possibility that an apparent reduction in infarct size observed early after the onset of reperfusion is subsequently overcome by delayed necrosis or apoptosis [14]. The infarct size was significantly smaller in the propofol and intralipid groups than in the control group, indicating that intralipid has a partial myocardial protective effect.
The role of cTn-I and cTn-T measurements in the diagnosis of patients with acute coronary syndrome is well established [15]. These measurements provide a definitive biochemical test to confirm or exclude myocardial necrosis. Troponin assays are not only more sensitive but are also more specific than a CK-MB assay. In this study, the I/R injury increased cTn-I serum levels, which were reduced by propofol infusion during the periischemic period.
This study had some limitations. First, the frequent lack of comparisons with a non-anesthetized control group is a difficulty when examining the effects of these intravenous anesthetic agents, particularly in vivo, because many studies have used acutely instrumented models requiring basal anesthesia [16-18]. Second, ketamine blocks the cardioprotective effects of ischemic preconditioning [19]. However, the effect is stereoselective, and R(-)-ketamine mainly blocks the effects of preconditioning, whereas racemic ketamine does not [20,21]. Racemic ketamine was used as the anesthetic drug in this study because it has the lowest myocardial protective effect compared to other anesthetic drugs. Third, the hemodynamic parameters were only checked at 48 h after reperfusion. They could not be checked at baseline or during the ischemic period due to technical difficulties.
In conclusion, a clinically relevant propofol concentration was associated with a delayed myocardial protective effect after a regional I/R injury in an in vivo rat heart model. This myocardial protective effect might not be due to delayed myocardial injury, but to the myocardial protective effect of propofol itself.
 XML Download
XML Download