

 PDF
PDF Citation
Citation Print
Print


Introduction
Because pretreatment with ischemic or pharmacological preconditioning is seldom possible in patients with ongoing acute myocardial infarction, strategies that directly target reperfusion could be more reliable. In this regard, ischemic postconditioning (I-Post) and pharmacological postconditioning (P-Post) have generated considerable interest recently. I-Post, which entails brief repetitive mechanical interruptions during early reperfusion, has reduced myocardial infarction in dogs [1], rodents [2], and even humans [3]. The infarct limitation effect by I-Post is as effective as that of ischemic preconditioning (I-Pre) [1]. In addition, P-Post with various adjuvants, such as opioid, bradykinin, insulin, and urocontin, has also been shown to provide cardioprotection [4-7].
N,N,N'N'-Tetrakis-[2-pyridylmethyl]-ethylenediamine (TPEN) is a high-affinity chelator of transition metals, such as Zn2+. TPEN also binds Ca2+ with a low affinity, and its fast diffusion across membranes renders it an intracellular Ca2+ buffer [8]. It has been proposed that ischemic pretreatment with TPEN confers cardioprotection in rat cardiomyocytes [9] and hearts [10], and TPEN treatment before and during hypoxia induces activation of a sarcolemmal Na+/Ca2+ exchanger and increases Ca2+ extrusion from the cytoplasm of cardiomyocytes, preventing cytosolic Ca2+ overload in cultured cardiomyocytes [9]. Furthermore, TPEN administered prior to ischemia has exerted beneficial effects on post-ischemic cardiac function; it has also reduced ventricular fibrillation (VF) incidence as well as the incidence and duration of ventricular tachycardia in in vivo rat hearts [10]. However, the TPEN induced myocardial infarct limitation effect, which is considered a more reliable endpoint than functional recovery [11], has not been characterized.
Interestingly, some drugs protect the myocardium against ischemic injury but not against reperfusion injury and vice versa [12-14]. In this regard, it should be determined whether TPEN also provides cardioprotection when administered during the reperfusion period, which is a more suitable management time in clinical settings. Thus far, however, there is scant information about targeting TPEN during the reperfusion phase and the subsequent effects it would have on infarct limitation and cardiac function recovery.
Therefore, in this study, we examined whether P-Post with TPEN at the concentrations that are cardioprotective against ischemia could reduce myocardial infarct size and induce cardiodynamic functional recovery in an isolated rat heart model.
Materials and Methods
Langendorff isolated heart perfusion preparation
Male Sprague-Dawley rats (280-330 gm) were obtained from Korea Taconic Company. Rats were anesthetized with 100 mg/kg of pentobarbital sodium (Entobar®, Hanlim Pharmacy, Korea), and 300 IU of heparin was administered intraperitoneally. Coronary perfusion by the Langendorff system with modified Krebs-Henseleit (KH) solution, containing 118.5 NaCl, 4.7 KCl, 1.2 MgSO4, 1.8 CaCl2, 24.8 NaHCO3, 1.2 KH2PO4, and 10 glucose (in mM), was performed as previously described in detail in our previous reports [15,16].
Making of ischemia and reperfusion
All hearts were allowed to stabilize for at least 20 min before ischemia induction. Hearts were subjected to 30 min of regional ischemia and 120 min of reperfusion. To induce regional ischemia, a snare was made at the level of the proximal length of the left coronary artery (LCA). Regional ischemia was induced by pulling the snare and confirmed by regional cyanosis and a substantial decrease in left ventricular developed pressure (LVDP). Reperfusion was started by releasing the snare. Hearts experiencing VF after reperfusion usually revert spontaneously to sinus rhythm. VF lasting more than 30 s was treated with finger flick cardioversion until a perfusing rhythm was obtained. No pharmacological agents were used for defibrillation.
Assessment of cardiac function
In isolated hearts, a KH buffer-filled latex balloon was inserted into the left ventricle (LV) and was adjusted to 5-10 mmHg of the left ventricular end-diastolic pressure (LVEDP) at the beginning of the experiment. Cardiodynamic variables, including heart rate (HR), left ventricular systolic pressure (LVSP), and LVEDP, were recorded with the MP150 system (BIOPAC Systems Inc., USA). LVDP and rate-pressure product (RPP) were calculated as follows: LVDP = LVSP - LVEDP and RPP = LVDP × HR. Maximum (+dP/dtmax) and minimum (-dP/dtmin) of the first derivative of LV pressure were analyzed using the Acqknowledge software (version 3.9.0.). Coronary flow (CF) was measured by perfusate dripping from the right heart into a graduated cylinder.
Experimental protocol
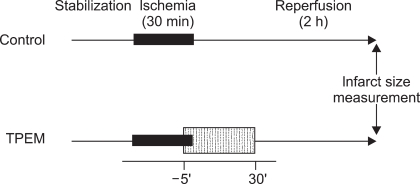
Isolated rat hearts were assigned randomly to one of the following two groups: 1) Control; no other intervention either before or after LCA occlusion (n = 9), and 2) TPEN; 10 µM of TPEN (n = 8). TPEN (Sigma-Aldrich Chemical, USA) was infused for a period of 5 min before and 30 min after reperfusion (Fig. 1). TPEN was dissolved with dimethyl sulfoxide and stored at -20℃. TPEN was diluted with KH solution to the required final concentrations, as described above, on the day of each experiment. The concentration of TPEN used in this study was based on a previous study for isolated working rat hearts [10].
Exclusion criteria
We decided prospectively that any hearts with a HR < 250 beats/min, those failing to develop LVSP > 80 mmHg when the LVEDP was kept at 5-10 mmHg, or CF > 18 ml/min or < 8 ml/min at the end of stabilization would be excluded from the study.
Determination of ischemic and infarct zone
At the end of each experiment (2 h reperfusion), ischemic (area at risk, AR) and infarcted (area of necrosis, AN) regions were measured with fluorescent polymer microspheres (Duke Scientific Corp., USA) and 2,3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich Chemical, USA) staining as described previously [15]. The AR and AN regions were quantified with the ImageTool program (UTHSCSA Image Tool, version 3.0). Infarct size was expressed as a percentage of the risk zone (AN/AR). All measurements were performed in a blinded fashion. Body weight (mean 302.2 ± 4.4 gm) and heart weight (mean 1.54 ± 0.04 gm) were equivalent between the two groups.
Results
All hearts for this experiment were perfused within 30 s after excision. A total of 19 rat hearts were used for the infarct size measurement study. Two hearts were excluded for the following reasons: HR < 250 beats/min (1) and CF > 18 ml/min (1) after the stabilization period. Therefore, we report the data for 17 hearts that successfully completed the infarct experiment study. The incidence of VF during the first 30 min after reperfusion was 67% (6/9) in the control group and 62% (5/8) in the TPEN group, respectively. Statistical analysis was not performed for the occurrence of VF, because of the small sample size. Hearts experiencing VF after reperfusion usually reverted to sinus rhythm spontaneously or by finger flick cardioversion.
Infarct size
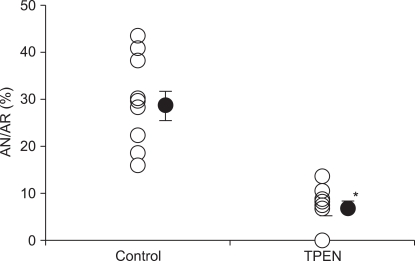
Fig. 2 represents the AN/AR ratio after 2 h of reperfusion by TTC stain. AN in control hearts was 29.5 ± 3.2% of the AR, which is in agreement with our recently reported infarct measurement study [15,16]. AN/AR of the TPEN group was significantly reduced (6.9 ± 1.7%) compared to control hearts (P < 0.001).
Functional recovery
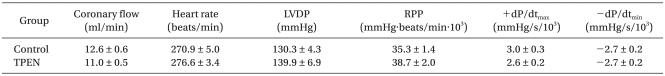
Baseline CF and HR after stabilization are shown in Table 1 and were comparable between the groups, averaging 11.8 ± 0.5 ml/min and 273.6 ± 3.1 beats/min, respectively. CF abruptly decreased after ischemia and diminished continuously during reperfusion without achieving statistically significant differences between the two groups. There was no statistically significant difference in HR throughout the experiment between the groups (data not shown).
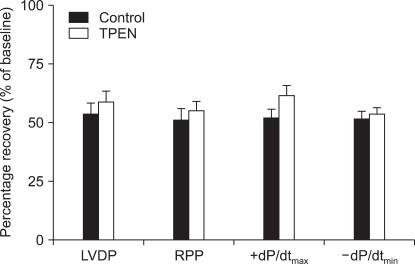
Baseline values of cardiodynamic parameters after stabilization are also shown in Table 1. There was no significant difference in baseline cardiodynamic parameters between the two groups (P > 0.05). Fig. 3 shows recovery of LVDP, RPP, +dP/dtmax, and -dP/dtmin after 2 h of reperfusion compared to baseline levels. There were no significant differences in cardiodynamic variables between the two groups (P > 0.05).
Discussion
TPEN is a drug that was proposed to protective against myocardial ischemic injury and reduce post-ischemic cardiac damage [17]. Until now, most of the studies on TPEN's cardioprotective effect were performed on ischemic pretreatment, and the effect of P-Post with TPEN remained. The present study has shown for the first time that targeting the use of TPEN, a metal chelator, during the reperfusion phase limits myocardial infarction. In addition, the study showed that there were no differences in cardiodynamic parameters, such as LVDP, RPP, +dP/dtmax, and -dP/dtmin, between TPEN-treated and non-treated hearts. Our data showed that P-Post with TPEN reduces myocardial infarct size but does not improve the functional recovery after reperfusion in isolated rat hearts.
Although tissue salvage by an anti-infarct intervention leads to improvement of cardiac function, infarct size limitation does not always produce mechanical functional recovery of hearts after myocardial ischemic/reperfusion (I/R) injury. For example, a potent opioid such as fentanyl, or angiotensin II reduces infarct size but does not protect against myocardial dysfunction in isolated rat hearts [18,19]. In addition, there are also several lines of evidence that I-Pre and I-Post also limit infarct size but do not reduce myocardial dysfunction [20,21]. The adenosine receptor agonist AMP579 or nitric oxide donors, on the other hand, alleviate both myocardial infarct size and dysfunction [22,23]. The discrepancy between infarct limitation and cardiac function recovery may be explained by continued stunning, or the possibility that the salvaged tissue is not yet normal [11]. Myocardial stunning is prolonged myocardial dysfunction with the histological absence of necrosis after I/R injury. It is proposed that stunning occurs in reversibly injured cells or via nonlethal injury to the epicardial border zones surrounding infarcted myocardium [24]. Although cellular ATP levels or free radicals have been proposed to be important mediators for stunning [25], the exact mechanism responsible for stunning remains unclear. Another possible mechanism for myocardial stunning is an increase in the concentration of intracellular Ca2+. The increase in the Ca2+ concentrations in myocardial cells is likely explains the depressed ability of I/R hearts to generate contractile force [26]. In our present study, targeting TPEN during the reperfusion period reduced myocardial infarction but did not attenuate post-ischemic systolic dysfunction, even though TPEN has a Ca2+ buffer action. We do not know the exact reason for this, but the Ca2+ buffer action of the TPEN concentration (10 µM) used during reperfusion might not be enough to attenuate myocardial systolic dysfunction.
It has been previously demonstrated that TPEN reduces basal cardiac nitric oxide (NO) content and prevents the accumulation of NO during myocardial I/R [9]. In addition it has been proposed that lowering sarcolemmal Ca2+ content may be a mechanism underlying the recently reported cardioprotective and anti-arrhythmic features of TPEN [9,10]. A chelator is a substance consisting of molecules that bind tightly to metal atoms, thus forcing the metal atoms to go wherever the chelator goes. Though TPEN has been used mainly as a heavy metal (e.g. Zn2+) chelator, it has a low affinity for Ca2+ as well and its fast diffusion across membranes make TPEN a membrane permeable buffer for Ca2+ within intracellular stores [8]. Recently, Shmist et al. [9] demonstrated that TPEN improves myocardial protection against ischemia by modulation of intracellular Ca2+ homeostasis. Jung et al. [27] also demonstrated that myocardial protection by TPEN has been related to its modulation of Ca2+ homeostasis through the activation of the Na+/Ca2+ exchanger leading to increased extrusion of Ca2+ from the cytoplasm in ventricular myocytes. However, the exact mechanism of the cardioprotective effect achieved by targeting the administration of TPEN during the reperfusion period has not yet been well established and should be determined in the future.
Interestingly, some drugs protect the myocardium when used during the ischemic phase but not in the reperfusion phase and vice versa [12-14]. In this study, administrating TPEN during the reperfusion phase effectively reduced myocardial infarction. Taken together with other previous reports on the effect of PEN treatments targeting ischemia [8,9], one can propose that TPEN has anti-ischemic and reperfusion injury protective effects.
It is also interesting that TPEN acts as a membrane permeable Zn2+ chelator, i.e., TPEN reduces intracellular Zn2+ concentrations. Zn2+ is essential in maintaining the structure and function of cell membranes, and it plays an important role in the maintenance of the activity of various enzymes [28]. Recently, several lines of evidence documented that exogenous Zn2+ may lead to cardioprotection against reperfusion injury [29,30]. In our study, TPEN administration during the reperfusion phase significantly reduced myocardial infarction, even though TPEN reduces intracellular Zn2+ concentrations. The reason for this is unclear, but we can propose that the Ca2+ buffering effect of TPEN may be a more important mechanism for the limitation of infarction than the ability to lower the Zn2+ concentration.
We conclude that P-Post with a 10 µM concentration of TPEN, which is proposed to have a cardioprotective effect during ischemia, limits myocardial infarction but does not attenuate myocardial dysfunction in isolated rat hearts. Further studies are needed to determine the exact mechanism for this discrepancy.
 XML Download
XML Download