

 PDF
PDF Citation
Citation Print
Print


Introduction
Liver injury caused by oxidative stress may occur under many clinical conditions, including liver surgery, transplantation surgery, the use of extracorporeal circulation in cardiac or vascular surgery and hemorrhagic, cardiogenic, or septic shock states followed by resuscitation [1,2], and it could increase the morbidity and mortality of patient [3,4]. Although hepatocytes have an antioxidant defence system to remove or neutralize reactive oxygen species (ROS), excessive production of ROS such as hydrogen peroxide (H2O2), occurring during the reperfusion period, results in an imbalance between pro-oxidants and antioxidants and usually leads to cellular dysfunction and tissue injury [5]. Thus, suppression of oxidative stress caused by ROS could reduce liver damage and ameliorate outcome of patients.
Propofol (2, 6-diisopropylphenol) and barbiturate, highly lipid-soluble anaesthetics, are both known to have antioxidant activities, protecting against lipid peroxidation [6], and both agents are often used in several clinical conditions to reduce cerebral edema during liver transplantation in fulminant hepatic failure (FHF) patients [7,8]. However, whether the antioxidant activities of propofol and pentobarbital protect hepatocytes exposed to oxidative stress is unclear.
Cell death can be categorized into apoptosis and necrosis. Apoptosis, or programmed cell death, is an active process character ized by cytoplasmic shr inkage, chromat in condensation, nuclear fragmentation, and activation of caspases [9]. In addition, phosphatidylserine (PS) is exposed on the external surface of the cell in the early phase of apoptosis, and this exposure precedes membrane damage and DNA fragmentation [10]. On the other hand, necrosis is passive, and is characterized by cell swelling, rupture of the plasma membrane, and cell lysis, with leakage of cytoplasmic components such as lactate dehydrogenase (LDH) [9].
It is known that direct exposure of hepatocytes to exogenous oxidants including H2O2 can induce both apoptotic and necrotic cell death [11]. In the current study, we explored the modes of cell death (apoptosis and/or necrosis) induced by H2O2 in hepatic cell and compared the protective effects of propofol and pentobarbital.
Materials and Methods
Propofol was obtained from AstraZeneca Pharmaceuticals (Macclesfield, Cheshire, UK), and pentobarbital sodium salt was from Sigma (St. Louis, MO, USA). The LDH assay kit (CytoTox 96® Non-Radioactive Cytotoxicity Assay) was from Promega (Madison, WI, USA). RPMI-1640 with L-glutamine, fetal bovine serum, and an antibiotic-antimycotic mixture (penicillin-streptomycin-amphotericin B) were purchased from Gibco BRL (New York, NY, USA). H2O2 was purchased from Sigma. Fluorescein isothiocyanate (FITC)-labeled Annexin V and propidium iodide (PI) were from BD Biosciences (San Jose, CA, USA).
The SNU-761 human hepatocellular carcinoma (HCC) cell line was purchased from the Korean Cell Line Bank [12]. Cells were cultured in RPMI-1640 with L-glutamine (300 mg/L) and HEPES (25 mM) containing 10% (v/v) fetal bovine serum and 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of amphotericin B at 37℃ in a humidified incubator under 95% air/5% CO2. Culture medium was replaced every 2 days. Three days after plating, cells were challenged with H2O2 at variable concentrations and different durations.
Varying concentrations of propofol (1, 10, 50 µM) or pentobarbital (10, 50, 100, 400 µM) were added to cell culture medium. Before induction of cell death by H2O2 (125 µM), cells were pretreated with different drug doses for 30 min and H2O2 was then added; incubation continued for 6 h.
LDH is normally present in the cytoplasm of hepatocytes. In response to cell damage, LDH is released from cells. Therefore, to measure necrotic cell death, we assayed LDH levels and calculated percentages of LDH release to the medium. LDH activity was measured spectrophotometrically using a commercial kit. The percentage of LDH release was calculated by [LDH]test/[LDH]control × 100 where [LDH]test was the LDH level of the test medium and [LDH]control was the measured LDH level in control medium.
To assess the cell death modes induced by H2O2, treated SNU-761 HCC cells were doubly stained with Annexin V-FITC and PI and analyzed by flow cytometry. Exposure of PS on the external cell surface occurs in the early phase of apoptotic cell death, during which time the cell membrane remains intact [13]. Therefore, simultaneous use of Annexin V (which binds specifically to PS) and a non-vital dye (such as PI; exclusion of the dye from cells indicates cell membrane integrity) is valuable in discriminating between apoptosis and necrosis [10].
Annexin V-FITC/PI double staining of cells was achieved using an Annexin V-FITC apoptosis detection kit. Briefly, both floating and adherent cells were collected and analyzed; 1 × 106 cells/well were plated on a 12-well plate. Cells were washed with PBS and collected by trypsinization. After centrifugation at 400 × g for 5 min at 4℃, cells were washed twice with cold PBS and resuspended in 1 x Annexin V binding buffer. One hundred microliter amounts of cell suspension were transferred to test tubes to which were added 5 µl of FITC-labeled Annexin V and 5 µl of PI solution. The cells were gently vortexed and incubated for 15 min at room temperature in the dark. After addition of 200 µl 1 x binding buffer to each tube, cells were analyzed on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). Annexin V-FITC and PI emissions were detected in the FL 1 and FL 2 channels. The Annexin V-FITC-/PI- population was considered to reflect normal healthy cells, whereas Annexin V-FITC+/PI- cells were taken to show early apoptosis. Annexin V-FITC+/PI+ cells were in late apoptosis or necrosis, whereas Annexin V-FITC-/PI+ cells were necrotic [11]. The percentages of normal, early apoptotic, late apoptotic, and necrotic cells were calculated using the CellQuest Pro software program (r 2000 Becton Dickinson, Hertfordshire, UK).
Statistical analyses were performed employing the SigmaStat program (version 3.10; Systat software; Chicago, IL, USA). Multiple groups were compared using one-way analysis of variance (ANOVA) or the Kruskal-Wallis one-way ANOVA on ranks followed by multiple comparisons using the Tukey post hoc test when appropriate. For all comparisons, a P value less than 0.05 was considered statistically significant. Data shown are mean ± SD unless stated otherwise.
Results
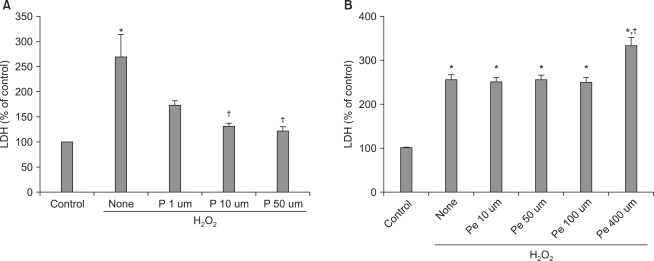
Pretreatment with propofol at 1, 10, or 50 µM gradually suppressed H2O2-induced LDH release, with the two higher concentrations showing significant release inhibition (P < 0.05) (Fig. 1A). Pretreatment with pentobarbital at 10, 50, or 100 µM did not suppress H2O2-induced LDH release; in fact, pentobarbital at 400 µM significantly (P < 0.05) increased such release (Fig. 1B).
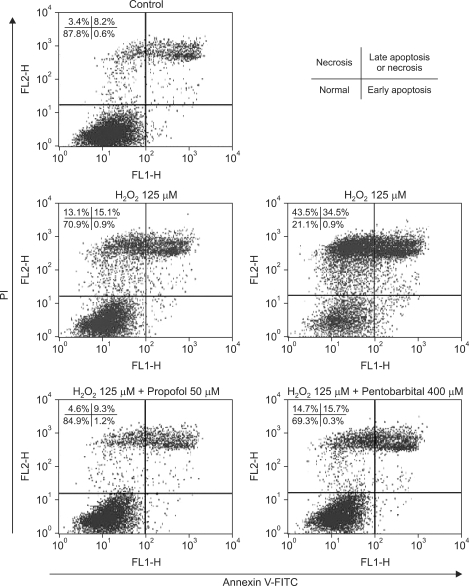
Annexin V-FITC binding analysis and PI staining were performed to identify cells undergoing apoptotic and necrotic death, respectively. SNU-761 HCC cells were treated with 0, 125, or 250 µM of H2O2 for 6 h and next stained with Annexin V-FITC/PI. As shown in Fig. 2, H2O2 decreased the proportion of normal cells, and increased the percentages of apoptotic and necrotic cells, in a dose-dependent manner.
After treatment of SNU-761 HCC cells with H2O2 for 6 h at a concentration of 125 µM, necrotic and late apoptotic cells increased in number. As shown in Fig. 2, propofol decreased these numbers (P < 0.05), but pentobarbital caused no significant decrease in numbers of necrotic or apoptotic cells.
Discussion
In this study, we examined and compared the effects of propofol and pentobarbital on hepatocyte death induced by H2O2 in vitro. The results indicate that propofol, but not pentobarbital, exerts a protective effect on hepatocytes exposed to H2O2 oxidant stress.
To investigate possible protective roles of propofol and pentobarbital during oxidant-mediated cell death, we used H2O2 as a stress inducer. ROS, including H2O2, cause a variety of cell and tissue injuries, and hepatocytes are susceptible to H2O2-induced damage. High levels of H2O2 probably overwhelm cellular antioxidant defenses, leading to cellular dysfunction and cytotoxicity [14]. H2O2 can induce apoptosis and necrosis in hepatocytes [15], and the relative proportions of the cell death modes may be determined by insult intensity and cell type [11]. We found that 6 h exposure to 125 µM H2O2 caused apoptosis and necrosis of SNU-761 HCC cells. To explore possible protective effects of propofol in cells under oxidative stress, we chose the range 1-50 µM (0.178-8.90 µg/ml) as clinically relevant because peak plasma concentrations of propofol are reportedly 40-60 µM (7.12-10.68 µg/ml) at anaesthesia induction and 10-25 µM (1.78-4.45 µg/ml) during anaesthesia maintenance [16,17]. Our results are not, however, in full agreement with previous studies, which yielded conflicting data regarding the effects of propofol on the liver under oxidative stress [18-20]. Navapurkar and co-workers [17] demonstrated that propofol could protect suspensions of isolated rat hepatocytes from an oxidant insult. Also, Wang and co-workers [20] reported that propofol had an anti-apoptotic effect on human hepatic L02 cells suffering from oxidant stress induced by H2O2; this was mediated by activation of extracellular signal-regulated kinase pathways. However, in an ischemia/reperfusion (I/R) model using the precision-cut rat liver-slice technique, propofol did not show any beneficial effect against I/R injury [19]. Reasons for these discrepancies may include the use of different experimental models. For example, liver slices are multi-layered, and include not only hepatocytes, but also other cell types (such as Kupffer cells and neutrophils) that inhibit the antioxidant effects of propofol [19].
In the present study, pentobarbital did not protect hepatocytes from oxidative stress; rather, this drug aggravated stress damage when present at a high concentration (400 µM). This concentration is comparable with the plasma level during barbiturate coma. Although central nervous system levels of pentobarbital during drug therapy are largely unknown, common therapeutic plasma concentrations have been reported to be 144-323 µM [21]. Therefore, we considered 1-400 µM of pentobarbital to be the clinically relevant range. Our results are consistent with those of a previous study showing that pentobarbital aggravates neuronal cell damage after combined oxygen and glucose deprivation [6]. Further work exploring the detrimental effects of pentobarbital may be warranted.
When the clinical implications of our data are considered, it may be that propofol is a better anesthetic choice than pentobarbital during surgery that involves temporary interruption of liver blood flow. In addition, pentobarbital has usually been employed to reduce cerebral edema during liver transplantation in FHF patients, but we may be more cautious attitude to use of pentobarbital in liver surgery. Delayed wakefulness and postoperative neurologic examination are caused by the very long drug half-life, and pentobarbital use is also associated with significant hemodynamic disturbances [7,8]. Therefore, some recent reports have suggested that propofol might be a useful alternative for reduction of cerebral edema because propofol metabolism is not greatly affected by liver failure [8,22]. In the present study, our data support the idea that propofol may be more useful than pentobarbital in patients with FHF. However, a further in vivo study is needed to establish which anaesthetics are suitable for use in hepatic surgery, particularly in FHF cases, during which interruption of liver blood flow is planned, because the in vitro and in vivo effects of anesthetics can differ [23]. Also it is another consideration that we used the tumor cells of human hepatocytes. In other words, different results might have made in our research if we had used normal human hepatocytes.
In conclusion, pentobarbital did not affect apoptosis or necrosis induced by H2O2, and, at low concentrations, was not cytotoxic for hepatocytes, whereas propofol preconditioning was protective in a dose-dependent manner. Our results show that propofol protects hepatocytes in vitro in the clinically relevant range 10-50 µM, suggesting potential benefits when the drug is employed during liver surgery, especially in FHF patients.
 XML Download
XML Download