

 PDF
PDF Citation
Citation Print
Print


Introduction
Telomerase reverse transcriptase (TERT) is a catalytic subunit of telomerase [1]. In normal tissues, the activity of telomerase is silenced, but when a single nucleotide variant (SNV) occurs in the two major hotspots [2] of the promoter region, telomerase is activated, which leads to telomere lengthening [3,4]. This series of processes is known to play a critical role in tumorigenesis in solid cancers [1,2,4], including gliomas [5,6].
In the 2016 World Health Organization (WHO) classification of central nervous system tumors, molecular parameters, including the status of the TERT promoter, are important for the integrated diagnosis of brain tumors [7]. When astrocytic gliomas harbor one of the following genetic variations: TERT promoter mutations, epidermal growth factor receptor (EGFR) amplification, or chromosome 7 gain/chromosome 10 loss, even though there is a lack of necrosis or microvascular proliferation, this tumor can be diagnosed as an isocitrate dehydrgenase (IDH)-wildtype glioblastoma, WHO grade IV [8]. Detecting the above molecular alterations in a very limited tumor sample would be the benefit the patients for important prognostic and potentially therapeutic information.
Next-generation sequencing (NGS) is one of the ideal methods for comprehensive molecular profiling of brain tumor. The NGS study can detect molecular alterations in wide genomic regions with relatively fast turnaround time, and low cost. However, the NGS study could yield plenteous information including inaccurate results, the appropriate approaches for extracting accurate information from NGS data would be a crucial step.
The promoter area of TERT has a high guanine-cytosine (GC) content (> 80%) and easily forms a secondary structure, such as a hairpin structure [9], resulting in a poor amplification. Therefore, many studies have suggested several ways to more accurately detect mutations in the TERT promoter region [10,11]. However, in the case of targeted NGS, which targets multiple genes, including TERT, the detection of variants in the TERT promoter region is often difficult due to poor amplification. In this regard, we planned to compare the results of NGS and Sanger sequencing of TERT and sought to find ways to accurately detect variants of the TERT promoter region using targeted NGS.
Materials and Methods
1. Case selection
We retrospectively selected 25 cases of IDH-wildtype glioblastoma from the NGS data in the pathology file of the Samsung Medical Center. All patients underwent brain tumor surgery. All patients provided informed consent for NGS. All tumors were histologically confirmed as glioblastomas, showing microvascular proliferation (n=25) or necrosis (n=22). Clinical information, such as sex, age, and tumor location, was collected. Tumor location was confirmed using preoperative magnetic resonance imaging. The mean age of the 25 patients was 63.64 years (range, 41 to 83 years; median, 65 years), and the male-to-female ratio was 14:11. The tumors were located in the cerebrum in 22 cases (frontal lobe in 10 cases, temporal in 6, parietal in 3, and frontoparietal in 3) and in the cerebellum in three cases. We also collected 10 anonymized non-neoplastic formalin-fixed paraffin-embedded (FFPE) stomach tissue samples for control.
2. DNA extraction
DNA was extracted from 5-μm-thick FFPE tissues using a QIAamp DSP DNA FFPE Tissue Kit (Qiagen, Hilden, Germany), and the extracted DNA was quantified using the QUBIT dsDNA BR Assay kit (Thermo Fisher Scientific, Waltham, MA).
3. Library preparation and targeted hybrid capture-based NGS
Target capture was performed according to the manufacturer’s protocol using the SureSelect XT automation reagent kit (Agilent, Santa Clara, CA), and a paired-end sequencing library was prepared using a barcode. The size and quality of the genomic DNA were validated using the Genomic DNA Analysis ScreenTape and Genomic DNA Reagent together with the Agilent 4200 Tape station (Agilent). Libraries were sequenced using the TG NextSeq 500/550 High Output Kit v2 (Illumina, Inc., San Diego, CA) and TG NextSeq 500/550 Mid Output Kit v2 (Illumina, Inc.).
4. Panel design and data analysis
We used a targeted sequencing panel pipeline named BrainTumorSCAN for data analysis, which was designed to cover 232 target genes at the Samsung Genome Institute. The list of 232 target genes is listed in S1 Table.
Paired-end reads were aligned to the human reference genome (GRCh37/hg19) using BWA-MEM v0.7.5 [12], SAMTOOLS v0.1.18 [13], GATK v3.1–1 [14], and Picard v1.93 (http://broadinstitute.github.io/picard/). SNVs were detected using MuTect ver. 1.1.4 [15], Lofreq ver. 0.6.1 [16], and VarDict ver. 1.06 [17] software. All of SNVs detected by each software were collected and merged. After collecting and merging, sequencing errors were filtered out through an in-house algorithm [18] and we also excluded variants which had been reported as benign or likely benign in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). Small insertions and deletions (Indels) < 30 bp in size were detected using Pindel v0.2.5a4 [19]. We used ANNOVAR for annotation of predicted SNVs [18]. Among all variants, genetic alterations with a variant of allele frequency (VAF) lower than 1%, a total read depth (TD) value lower than 50, or a variant read count (VC) value lower than 4 were considered as spurious variants and were excluded. To identify the somatic copy number alterations (CNAs), we calculated the mean read depth of each exon and normalized it according to the depth of the target regions in that sample. This normalized read depth was further standardized by dividing it by the expected read depth of a normal population. The expected read depth at each exon was taken from the median value of the read depth at that exon across a set of normal individual samples. Then, the amplitude of copy numbers was calibrated based on the calculated purity of the tumor cells in the sample for ratiocinating accurate copy numbers. If the calibrated amplitude of the copy number was greater than 4, it was considered as amplification, and if it was lower than 1, it was considered a deletion. Additionally, in case of Log2 scale of the adjusted copy number fold change in chromosomal short or long arm entirely is greater than 1 or lower than −1, it is considered as chromosomal short arm, long arm, or whole chromosomal gain or loss, respectively.
5. Sanger sequencing for TERT
Sanger sequencing for the analysis of TERT promoter mutation was performed using the following pair of primers as previously reported [20]. Amplification of the genomic DNA was performed using the GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA) and C1000 Touch Thermal Cycler kit (Bio-Rad, Hercules, CA), according to the manufacturer’s instructions. Sanger sequencing was performed using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems). The sequencing reaction was performed both in the forward and reverse direction, and the result was confirmed only when the forward-primer and reverse-primer results were consistent with each other.
6. O-6-methylguanine-DNA-methyltransferase (MGMT) promoter methylation analysis
A total of 2 μg of DNA was denatured using sodium hydroxide and modified using sodium bisulfite. Primer pairs specific for methylated, 5′-TTTCGACGTTCGTTCGTAGG-TTTTCGC-3′ (sense) and 5′-GCACTCTTCCGAAAACGAA-ACG-3′ (antisense), and unmethylated MGMT promoter, 5′-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3′ (sense) and 5′-AACTCCACACTCTTCCAAAAACAAAACA-3′ (antisense), were prepared. Control DNA for methylated and unmethylated samples was obtained from Qiagen control DNA. The PCR conditions used were as follows: 95°C for 10 minutes for 40 cycles, and 94°C for 15 seconds, 58°C for 30 seconds, 72°C for 30 seconds (35 cycles), and 72°C for 5 minutes. Electrophoresis for each PCR product was performed using an 8% acrylamide gel and ethidium bromide staining, and the gels were visualized under ultraviolet illumination.
7. Statistical analysis
All statistical analyses were performed using the SPSS software ver. 24.0 (IBM Corp., Armonk, NY). The general characteristics and demographic parameters were compared using chi-square and Fisher exact tests, and other quantitative data were analyzed using paired t tests. For comparisons between non-normally distributed variables, we used Shapiro-Wilk test and Mann-Whitney U test. A p-value lower than 0.05 was considered statistically significant.
Results
1. Detection of TERT promoter mutations using targeted NGS and Sanger sequencing
We reviewed the targeted NGS results for 25 glioblastomas. The overall sequencing quality and recommended guideline quality thresholds are summarized in S2 Table adopted from previous study [18]. The average Q30 values of the reads were 92.9% (range, 91.5% to 93.2%) and the on-target rate was 94.2% (range, 93.1% to 95.5%). The average TD value was 852.87±91.16 (range, 736.75 to 1,051.0; median, 839.58) and the average rate of uniformity was 78.0% (range, 74% to 81%).
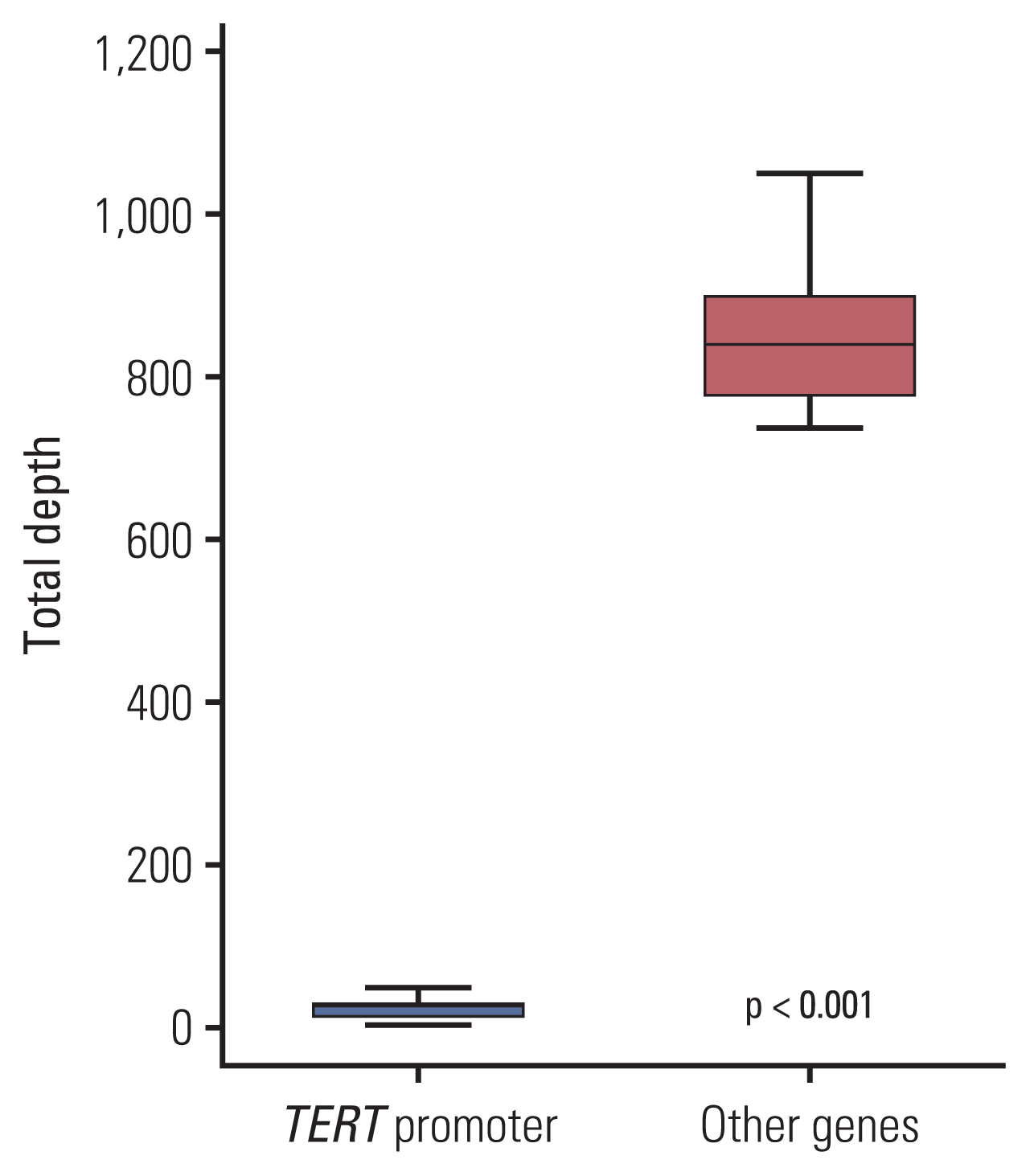
None of the tumors showed any mutations in the TERT promoter using targeted NGS, while TERT promoter mutations were detected in 17 out of 25 cases (68%) using Sanger sequencing (Table 1). To explore the different detection rates of mutations in the TERT promoter using NGS and Sanger sequencing, the aligned reads were manually checked using an integrative genomics viewer (IGV) (http://software.broadinstitute.org/software/igv/), considering the possible filtering because of the unsuccessful amplification of the TERT promoter. When reviewing the results of the analysis using an IGV (Fig. 1), we found TERT mutations in 16 of 25 cases (64%), and their TD, VC, and VAF values are summarized in Table 1. One case (case 7) showed a TERT promoter mutation in Chr 5:1,295,228 using Sanger sequencing, but the TERT promoter region could not be amplified using targeted NGS. As shown in Table 1, this tumor showed a very low read depth (2) in that position. The average TD value of the TERT promoter region was 21.59 (range, 2 to 47; median, 25) and was significantly lower than the average TD values of other genes (mean, 885.40; range, 736.8 to 1,051.0; median, 839.6) (p < 0.001) (Fig. 2). The average VC value of the TERT promoter mutation was 7.18±6.5 (range, 0 to 28; median, 6), and the average VAF value was 32.2±18.4 (range, 0 to 75; median, 28.6). All cases were excluded for the TERT promoter mutation in the original NGS study because of the auto-filtering criteria.
2. Modification of baits design to detect TERT mutations
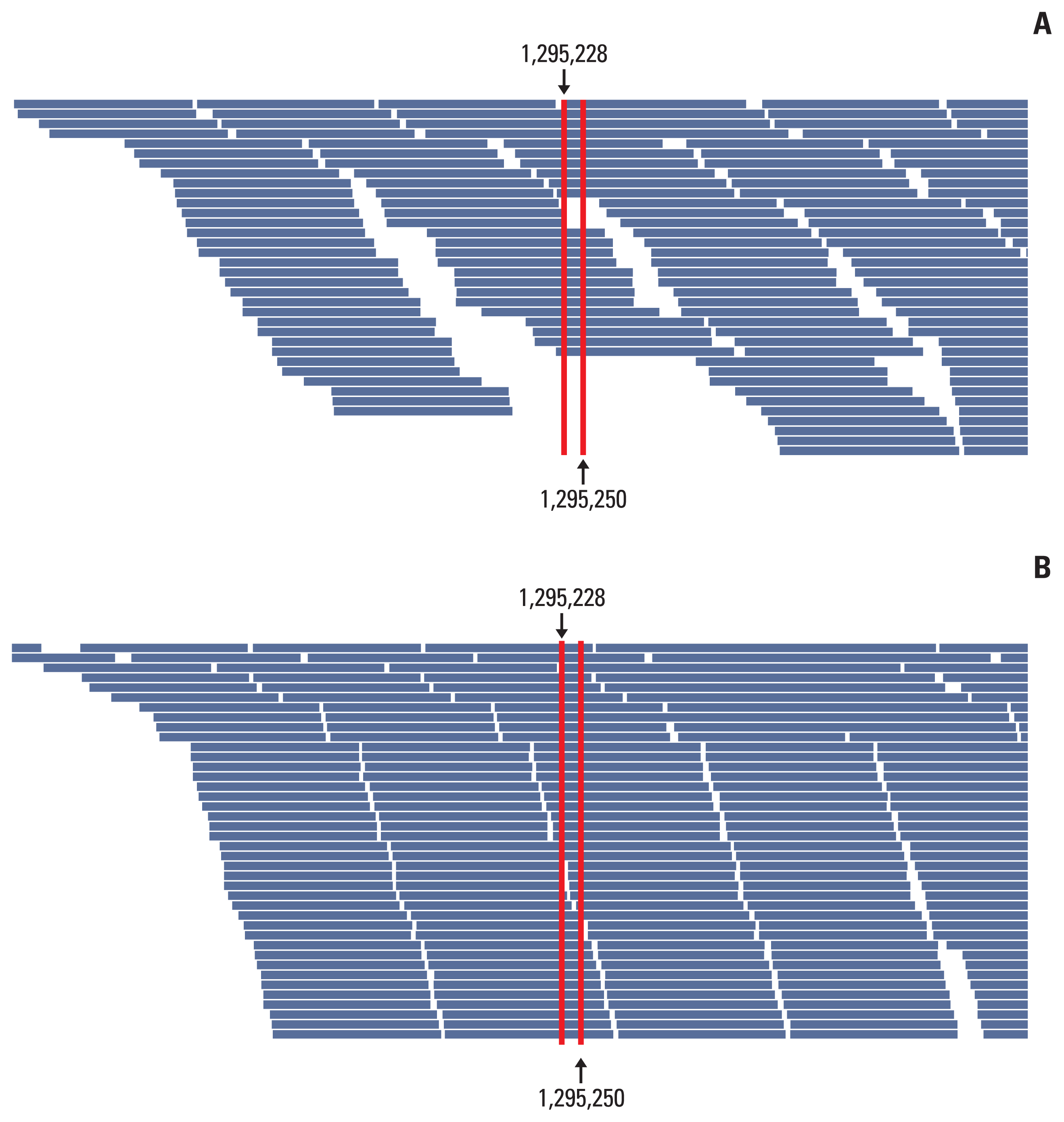
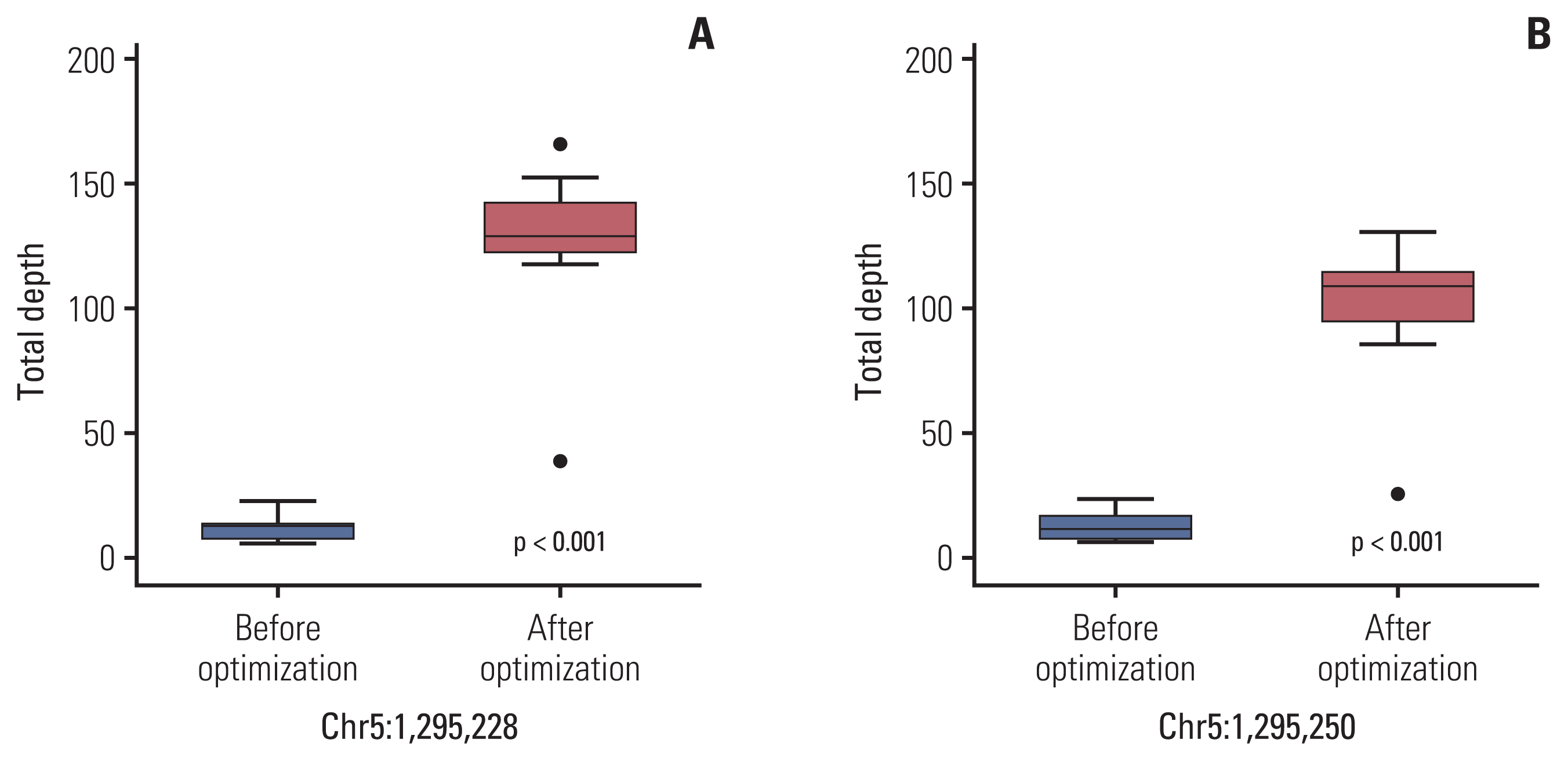
To improve the TD in the TERT promoter area, we corrected the design of the capture probe, the so-called baits, which were the single-stranded oligonucleotides used for hybridization with the DNA or RNA in the target capture process. We increased the number of baits around the TERT promoter region, especially in two hotspot areas, for increasing the coverage of these baits, and extended the area of the overlapped regions covered by the baits. Then, targeted NGS was performed on 10 cases of non-neoplastic gastric tissues. The results of the analysis of these cases using the IGV showed a change in the distribution of baits around the TERT promoter region (Fig. 3). The mean read depth of the two hotspots (Chr 5:1,295,228 and Chr 5:1,295,250) where mutations occur predominantly in the TERT promoter region were originally 12.3 and 13.1, respectively. After the modification of the baits in the TERT promoter region, the mean read depth of the two hotspots increased significantly to 124.89 and 99.2 (p < 0.001), respectively (Fig. 4). The mean Phred quality score (before modification, 33.95; after modification, 31.7) were not significantly different (p > 0.05) and the mapping quality rate score was 60 in all cases, before and after.
3. Genetic landscape of glioblastomas
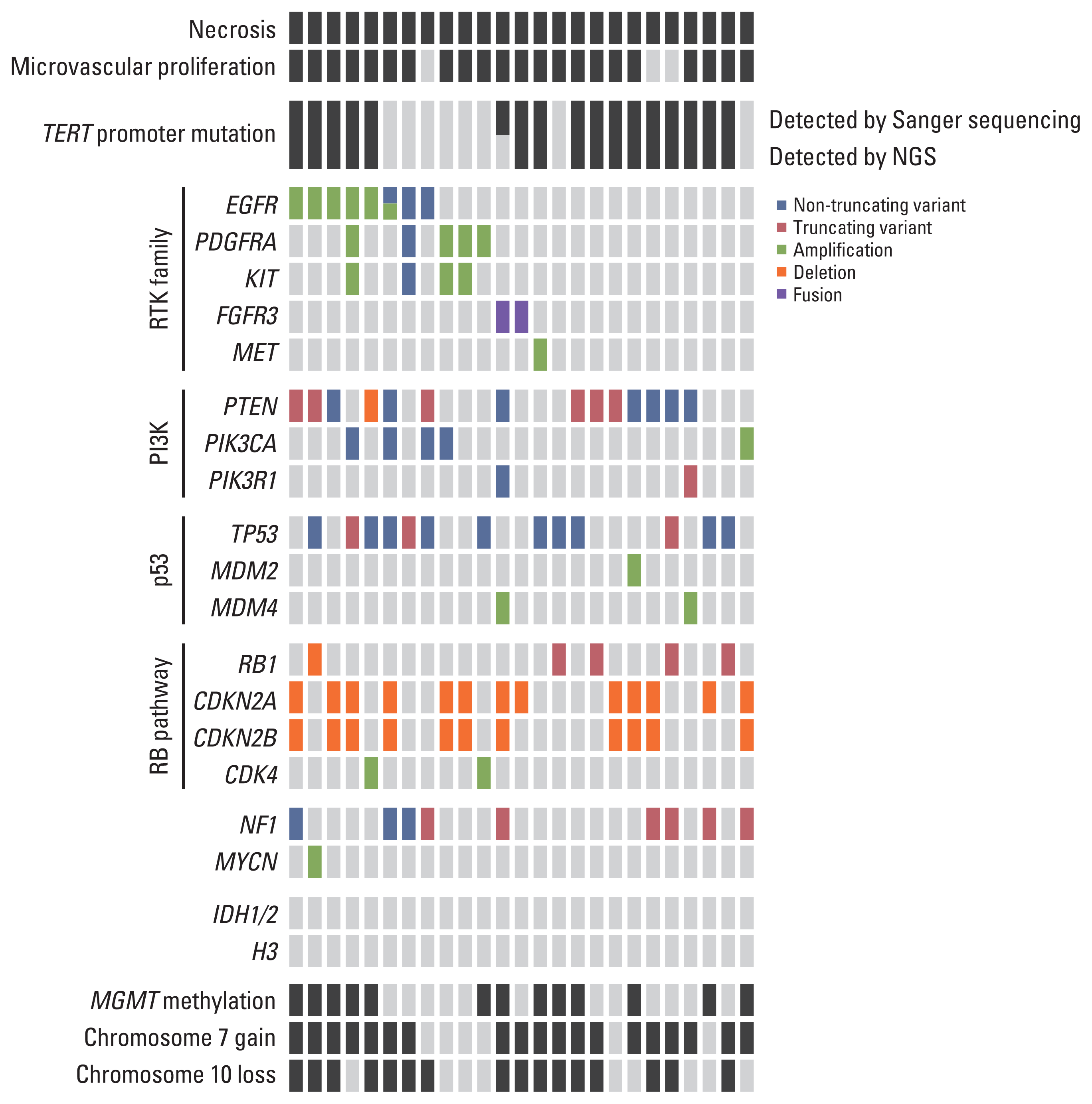
The NGS mutational analysis and Sanger sequencing detected 328 genetic alterations, including 201 non-synonymous SNVs, 36 truncating variants (stop-gain, frameshift, or splice-disrupting variant), seven in-frame indels, 82 CNAs, and two fusions (S3 Table).
Seventeen out of 25 cases (68%) harbored a TERT promoter mutation. Whole chromosome 7 gain was detected in 19 cases (76%), and whole chromosome 10 loss was detected in 14 cases (64%). MGMT promoter methylation was found in 56% of the cases. Six out of 25 cases (24%) showed EGFR amplification. The other genetic alterations observed are shown in Fig. 5. The landscape of glioblastomas evaluated in our study was not significantly different from those of other studies.
Discussion
As the diagnostic criteria for brain tumors have been more detailed and molecular techniques for their diagnosis have been developed, various genes related to their prognosis have come into the spotlight, such as TERT [10,12] or EGFR [21]. The detection of alterations in these genes can be used to predict a worse prognosis and to choose an appropriate treatment strategy and increase the survival of patients with gliomas.
In particular, recurrent mutations in two sites of the TERT promoter region have been found in Chr 5:1,295,228 and Chr 5:1,295,250, which are referred to as C228T and C250T, respectively. About 72%–90% of glioblastomas [3,6] and 95% of oligodendrogliomas [21,22] show a mutation of the TERT promoter in these two hotspots. Among histologically proven WHO grade II or III IDH-wildtype diffuse astrocytic gliomas, tumors with genetic alterations (EGFR amplification, +7/−10, TERT promoter mutations) are associated with an aggressive clinical behavior and show DNA-methylation profiles similar to those of IDH-wildtype glioblastomas [21,22]. In line with this, cIMPACT-NOW announced that for IDH-wildtype gliomas with TERT promoter mutations, EGFR amplification or whole chromosome 7 gain/10 loss was predicted to show a prognosis similar to that of glioblastomas and could be classified as a glioblastoma [8]. Therefore, the evaluation of TERT promoter mutations can be a critical step in the diagnosis of high-grade astrocytic tumors.
However, the TERT promoter region contains a GC-rich region [10] and forms a secondary structure, such as a hairpin structure, during PCR. It is usually amplified unsuccessfully and may show false-negative results [9]. Therefore, numerous approaches have been attempted for the appropriate amplification of genes having GC-rich regions: modification of the cycling environment, such as the temperature or cooling time [23,24]; the addition of various organic molecules, including dimethylsulfoxide [25]; or increasing the concentration of chemicals important to the amplification process, such as MgCl2 [26].
In high-throughput sequencing, the read depth of the GC-rich region is significantly lower than that of other regions [27], and has been a chronic problem in NGS data analysis. The unimodal GC curve shows that the read depth of GC-poor or GC-rich fragments is significantly lower than the average read depth [27]. Hybrid capture-based NGS is expected to be more vulnerable to GC bias due to an unexpected behavior in the solution-hybridization process [28].
In the present study, TERT mutations were initially not detected using targeted NGS, but were detected using Sanger sequencing, and to explore the different detection rates of mutations in the TERT promoter using NGS and Sanger sequencing, we checked the filtering guidelines of our targeted NGS. We found variants, but they showed TD values lower than 50 or VCs lower than 4. These variants were removed by our filtering guidelines to exclude fake variants caused by sequencing errors. These results were similar to those of a previous study by Sahm et al. [29]. They also reported that calls from the TERT promoter position were filtered because the average read depth of the hotspot of the TERT promoter was low. The present study demonstrated that a manual review of the aligned reads using the IGV showed a TERT promoter mutation in 68% of the 25 cases. Therefore, given that a poor read depth due to GC bias in the GC-rich region is a problem, it is necessary to manually check the reads directly without relying on filtering. Besides a manual review of the aligned reads, there are other methods that can be used to minimize the GC bias. To compensate for the poor amplification of GC-rich sequences, we attempted to expand the target area of the TERT promoter region captured through baits. During the process of target capture, the baits hybridize with genomic targets of interest and select target sequences from DNA libraries for amplification. Therefore, by designing baits of the TERT promoter region more tightly and increasing the number of baits targeting the TERT promoter region, the probability of the amplification of the targeted region was increased to reach a meaningful quality for amplification.
In addition to the GC content, several factors may be associated with biases in the NGS analysis. Chen et al. [30]reported that DNA synthesis bias and PCR stochasticity bias impacted the NGS bias to a greater extent than the GC bias. They also introduced a computational model predicting the molecular bias produced by the DNA synthesis and PCR sequencing retrieval process [30].
This study is signifying because it proposes an enforceable solution different from those presented in previous studies, such as increasing the amount of sequencing data or maintaining the ‘fragment length’ of DNA data [27]. It will also benefit many institutions that are preparing to design customized NGS panels for patients with brain tumors. In addition to TERT, the first exon of genes and the 5′ untranslated region or promoter region containing GC-rich regions [31] can provide important clinical information for patients.
In conclusion, we reaffirmed the harmful influence of GC bias in the interpretation of NGS data and provided a strategy for optimizing the NGS data analysis. Although high-throughput NGS provides abundant data, the underestimation of molecular bias can lead to the obsolescence of major portions of these data. Therefore, it is crucial to keep in mind the importance of molecular bias and to carefully interpret NGS data.

 XML Download
XML Download