

 PDF
PDF Citation
Citation Print
Print


Introduction
Triple-negative breast cancer (TNBC) is a subtype of breast cancer defined by the negative expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) [1,2]. Although TNBCs account for approximately 15–20% of all the breast cancer cases, they are the most challenging as they confer a poor survival outcome due to the limited number of available therapeutic options [1,3]. Since TNBCs lack ER, PR, and HER2, chemotherapy occupies the central position in the treatment of TNBCs. Anthracycline- and taxane-based chemotherapy have been the core of the chemotherapy regimens used for TNBCs [3–5]. However, the patients with TNBC still have very limited chemotherapy options compared with the patients with non-TNBC subtypes after tumor recurrence and metastasis, with a median response duration of only 3 months [3–5]. Furthermore, since anthracycline-based regimens have cardiotoxicity, it is essential to develop a novel regimen or to appropriately combine treatment regimens to limit the dose of anthracycline-based regimens [6,7].
As the therapeutic target of TNBCs, the mammalian target of rapamycin (mTOR) protein kinase attracts attention because it lies at the center of TNBC signal transduction [6,8]. Among the mTOR inhibitors, everolimus, an allosteric mTOR complex 1 (mTORC1)-specific inhibitor, has been approved for clinical use against ER positive breast cancer with exemestane (an aromatase inhibitor), and is currently in clinical trial against TNBC in combination with other chemotherapy regimens [6]. mTOR consists of two distinct cell signaling complexes, mTORC1 and mTORC2, both of which are essentially involved in cell proliferation. Therefore, targeting to mTORC1 alone may result in drug resistance due to compensatory activation of mTORC2 signaling [8–10]. Therefore, dual mTORC1 and mTORC2 inhibitors could improve the therapeutic efficacy of mTOR-targeted treatment by eliminating feedback activation of Akt. Ku-0063794 is a highly specific ATP competitive mTOR inhibitor that targets both mTORC1 and mTORC2 complexes [8]. Ku-0063794 inhibits the phosphorylation of S6K1 and 4E-BP1, which are downstream substrates of mTORC1, and also inhibits Akt phosphorylation on Ser473, which is the target of mTORC2 [8]. Based on these findings, we hypothesized that a combination of Ku-0063794 with docetaxel (a taxene-based regimen), could have enhanced anticancer activity against TNBCs. Thus, we investigated the anticancer effects and mechanism of Ku-0063794 and docetaxel treatment against TNBC cells in vitro and in vivo.
Go to : 
Materials and Methods
1. Chemicals
Ku-0063794 was purchased from Selleckem (Huston, TX). Docetaxel was purchased from Sigma Aldrich (St. Louis, MO).
2. Cell culture
MCF7 breast cancer cell line and MDA-MB-231 TNBC cell line were obtained from Korean cell line bank (KCLB, Seoul, Korea). MCF7 and MDA-MB-231 cells were maintained in RPMI (Thermo Fisher Scientific, Carlsbad, CA), supplemented with 10% fetal bovine serum (GibcoBRL, Carlsbad, CA) and 1% penicillin-streptomycin (Thermo Fisher Scientific, Carlsbad, CA), at 37°C in a humidified atmosphere with 5 % CO2 in an incubator.
3. Cell viability assay
Viability of MCF7 and MDA-MB-231 breast cancer cells were evaluated using EZ-Cytox Cell Viability Assay kit (Itsbio, Seoul, Korea) according to the manufacturer’s instructions.
4. Western blot analysis
MCF7 and MDA-MB-231 breast cancer cell lines were lysed using the EzRIPA Lysis kit (ATTO Corporation, Tokyo, Japan), and quantified by Bradford reagent (Bio-Rad, Hercules, CA). Proteins were visualized by western blot analysis using the primary antibodies (1:1,000 dilution) and then with HRP-conjugated secondary antibodies (1:2,000 dilution) from Vector Laboratories (Burlingame, CA). Specific immune complexes were detected using the Western Blotting Plus Chemiluminescence Reagent (Millipore, Bedford, MA). Primary antibodies against poly-ADP(adenosine diphosphate)-ribose polymerase (PARP), myeloid cell leukemia 1 (Mcl-1), E-cadherin, vimentin, snail, microtubule-associated proteins 1A/1B light chain 3B (LC3B), and p62 were obtained from Cell Signaling Technology (Beverly, MA). β-Actin was obtained from Sigma Aldrich.
5. Quantification of apoptotic cell death by flow cytometry
To detect apoptotic cell death, MCF7 and MDA-MB-231 breast cancer cells were stained with annexinV/propodium iodide (PI). After incubation for 10 minutes in the dark at 25°C, the cells were analyzed using Attune NxT acoustic focusing cytometer (Thermo Fisher Scientific, Waltham, MA).
6. Cell migration assay
MCF7 and MDA-MB-231 breast cancer cell line migration was analyzed using the in vitro wound healing assay. Cells were grown to confluence in 6-well plates and changed to serum-free medium for an additional 24 hours. Cell monolayers were scraped with a micropipette tip and treated with Ku-0063794, docetaxel or a combination of both agents. The wound area was photographed using phase-contrast microscopy before and 24 hours after the treatment. The percentage of wound closure was determined as: [(initial area–final area)/initial area]×100 [11].
7. Immunofluorescence and immunohistochemistry
MCF7 and MDA-MB-231 breast cancer cell lines were cultured on Lab-Tek chamber slides (Thermo Fisher Scientific). The cells were washed three times with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 20 minutes, and permeabilized with 0.3% Triton X-100 for 10 minutes. After blocking with 0.2% bovine serum albumin for 1 hour at 25°C, the slides were incubated with the antibodies against E-cadherin, vimentin, snail, LC3B, p62, and glyceraldehyde 3-phosphate dehydrogenase (1:200 dilution) at 4°C overnight. The slides were washed three times with PBS and incubated with Alexa Fluor 488– or Alexa Fluor 594–conjugated secondary antibodies (1:500 dilution) for 1 hour at 25°C; the nuclei were counter-stained with DAPI-containing VECTASHIELD Mounting Medium (Vector Laboratories) for 1 minute. The samples were observed using a fluorescence imaging system (EVOS U5000, Invitrogen, Carlsbad, CA) to analyze the expression of these markers.
For immunohistochemistry, paraffin-embedded tissue sections were deparaffinized in xylene and rehydrated in a series of graded ethanol. The antigen was retrieved with 0.01 M citrate buffer (pH 6.0) by heating the sample in a microwave. The tissue sections were then placed in 3% hydrogen peroxide for 5 min to inactivate the endogenous peroxidase, blocked for 10 minutes with normal horse serum (Vector Laboratories) and incubated with the primary antibodies against cleaved caspase-3 and Bcl-xL overnight at 4°C. The slides were then treated with the biotinylated secondary antibody for 30 minutes at 25°C, followed by streptavidin-HRP and 3,3′-diaminobenzidine solution for another 10 minutes at 25°C.
8. In vivo xenograft model
BALB/c nude mice (5 weeks) were used for comparative modeling of subcutaneous tumor growth of MCF7 and MDA-MB-231 cells. Breast cancer cells (5×106) were subcutaneously injected into each mouse. Fourteen days after tumor cell injection, all mice had measurable tumors. Mice were then randomly grouped (n=5 per group) and treated intraperitoneally with normal saline (control), Ku-0063794 (1 mg/kg in 100 μL normal saline, 3 times a week), docetaxel (1 mg/kg in 100 μL in normal saline, 3 times a week), and a combination of both agents (1 mg/kg Ku-0063794 combined with 1 mg/kg docetaxel in 100 μL normal saline, 3 times a week) for 3 weeks. Tumor size was measured twice weekly using a caliper. After the completion of treatment, all mice were euthanized.
9. Statistical analysis
All data were analyzed with SPSS ver. 11.0 software (SPSS Inc., Chicago, IL), and are presented as mean±standard deviation. Statistical comparison among the groups was performed using Kruskal-Wallis test. Probability values of p < 0.05 were regarded as statistically significant.
Go to :
Results
1. Cell viability after docetaxel and Ku-0063794 mono- and combination therapy
First, the effects of mono- and combination therapy comprising Ku-0063794 and docetaxel on the viability of MCF7 and MDA-MB-231 cells were investigated (Fig. 1). Docetaxel and Ku-0063794 monotherapies were shown to reduce the viability of these cells in a concentration- and time-dependent manner. The effects of docetaxel and Ku-0063794 combination therapy were different according to the cell type. Following the Ku-0063794 and docetaxel combination therapy, MDA-MB-231 cells showed the significantly decreased cell viability at lower concentrations than MCF-7 cells (p < 0.05). It suggests that the combination therapy has a synergistic effect on MDA-MB-231 cells rather than MCF-7 cells.
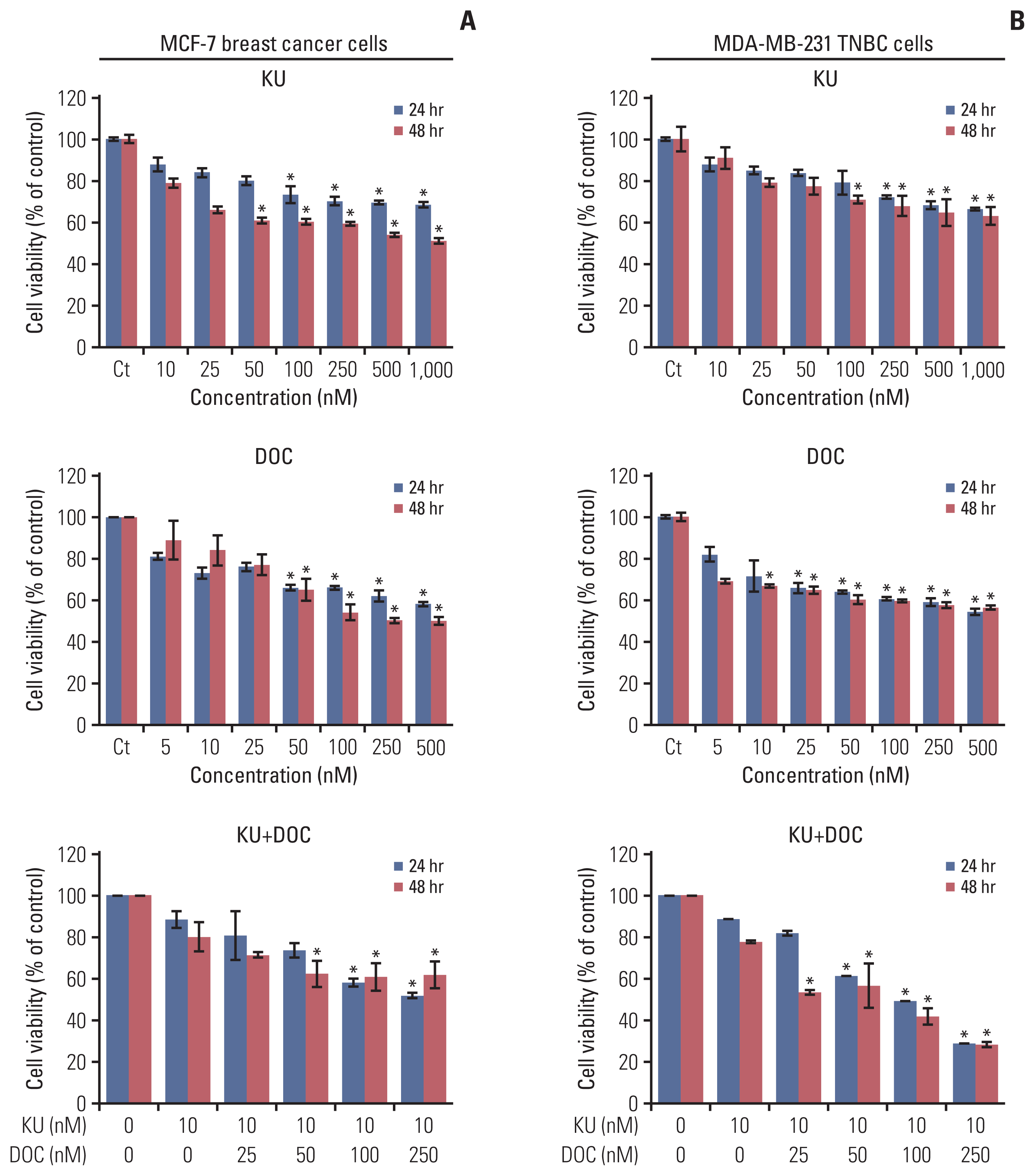 | Fig. 1Cell viability tests of breast cancer cells following docetaxel and Ku-0063794 mono- and combination therapy. (A) Viability of MCF-7 human breast cancer cells according to increasing concentration of Ku-0063794, docetaxel, and their combination. (B) Viability of MDA-MB-231 triple-negative breast cancer (TNBC) cells according to increasing concentration of Ku-0063794, docetaxel, and their combination. DOC, docetaxel; KU, Ku-0063794. Values are presented as mean±standard deviation of three independent experiments. *p < 0.05.
|
2. Apoptosis following mono- and combination therapies
The effects of mono- and combination therapy on cell apoptosis of MCF7 and MDA-MB-231 cells were determined using western blot analysis and flow cytometry, respectively. Western blot analysis revealed that combination therapy increased the expression of PARP (a pro-apoptotic marker) and decreased the expression of Mcl-1 (an anti-apoptotic marker) with the increasing concentration of docetaxel in the combination therapy (Fig. 2A). This pattern was not significantly different between the MCF-7 and MDA-MB-231 cells (Fig. 2B). Subsequently, Annexin V/PI double staining was used in flow cytofluorimetric analyses to detect apoptotic cell death (Fig. 2C and D). In both the cell groups, the population of Annexin V–positive cells (early and late apoptotic cells) tended to increase with the increasing concentration of docetaxel in the combination therapy, and this trend was more pronounced for the MDA-MB-231 cells than for the MCF-7 cells.
 | Fig. 2Cell apoptosis following docetaxel (DOC) and Ku-0063794 (KU) mono- and combination therapies. (A) Western blot analysis showing the effects of mono- and combination therapy on apoptosis of MCF7 breast cancer cells. Relative densities of individual markers. The relative densities had been quantified using Image J software and then were normalized to the density of β-actin in each group. (B) Western blot analysis showing the effects of mono- and combination therapy on apoptosis of MDA-MB-231 triple-negative breast cancer (TNBC) cells. Relative densities of individual markers. (C) Quantitative analysis of the effects of KU and DOC combination therapy on the apoptosis of MCF-7 breast cancer cells using Annexin V/propodium iodide (PI) staining and flow cytometry. Apoptotic cells were expressed as the total percentage of Annexin V-positive/PI-negative cells. (D) Quantitative analysis of the effects of KU and DOC combination therapy on the apoptosis of MDA-MB-231 TNBC cells using Annexin V/PI staining and flow cytometry. The total percentage of Annexin V–positive/PI-negative cells. Mcl-1, myeloid cell leukemia 1; PARP, poly-ADP (adenosine diphosphate)-ribose polymerase. Values are presented as mean±standard deviation of three independent experiments. *p < 0.05.
|
3. Epithelial-mesenchymal transition and cell migration following mono- and combination therapies
Western blot analysis was performed to determine the effects of mono- and combination therapy on the expression of the epithelial-mesenchymal transition (EMT)–related markers in the MCF7 and MDA-MB-231 cells. In the MCF-7 cells, individual monotherapies decreased the expression of E-cadherin (an epithelial marker) and snail (a mesenchymal marker) and increased the expression of vimentin (a mesenchymal marker), suggesting a tendency of EMT promotion (Fig. 3A). By contrast, combination therapy increased the expression of E-cadherin and decreased the expression of vimentin and snail compared with the expression of these markers following individual monotherapies, suggesting the effect of ameliorating EMT by combination therapy. This trend was more pronounced in MDA-MB-231 TNBC cells (Fig. 3B). Subsequently, we investigated cell migration following either mono- or combination therapies (Fig. 3C and D). In both types of cell lines, combination therapy was found to significantly reduce cell migration as compared to the individual monotherapies (p < 0.05).
 | Fig. 3Effects of docetaxel (DOC) and Ku-0063794 (KU), either individually or in combination, on epithelial-mesenchymal transition (EMT) and migration of breast cancer cells. (A) Western blot analyses showing the expression of EMT-related markers in MCF-7 breast cancer cells following mono- and combination therapies of DOC and KU. Relative densities of individual markers. The relative densities had been quantified using Image J software and then were normalized to the density of β-actin in each group. (B) Western blot analyses showing the expression of EMT-related markers in MCF-7 and MDA-MB-231 triple-negative breast cancer (TNBC) cells following mono- and combination therapies of DOC and KU. Relative densities of individual markers. (C) Wound healing assay (×200, scale bar=20 μM) showing the effects of DOC and KU, either individually or in combination, on the migration of MCF-7 and MDA-MB-231 breast cancer cells. Migration was expressed as percentage of cells migrated compared to the control. Values are presented as mean±standard deviation of three independent experiments. *p < 0.05.
|
4. Changes in autophagic markers following mono- and combination therapies
Autophagy induction leads to the upregulation of LC3B and downregulation of p62. We thus compared the expression of autophagy markers, LC3B and p62, in each of the cell types following docetaxel and Ku-0063794 treated either individually or in combination using western blot analysis. In the MCF-7 cells, individual monotherapies resulted in the higher expression of LC3B and lower expression of p62 as compared to the controls, suggesting autophagy induction (Fig. 4A). However, combination therapy resulted in a significantly lower expression of LC3B and higher expression of p62 as compared to the controls, suggesting autophagy inhibition (p < 0.05). In MDA-MB-231 cells, the upregulation and downregulation of LC3B and p63 was remarkable following docetaxel monotherapy (Fig. 4B). As with the MCF7 cells, combination therapy resulted in the significantly lower expression of LC3B and higher expression of p62, suggestive of autophagy inhibition (p < 0.05).
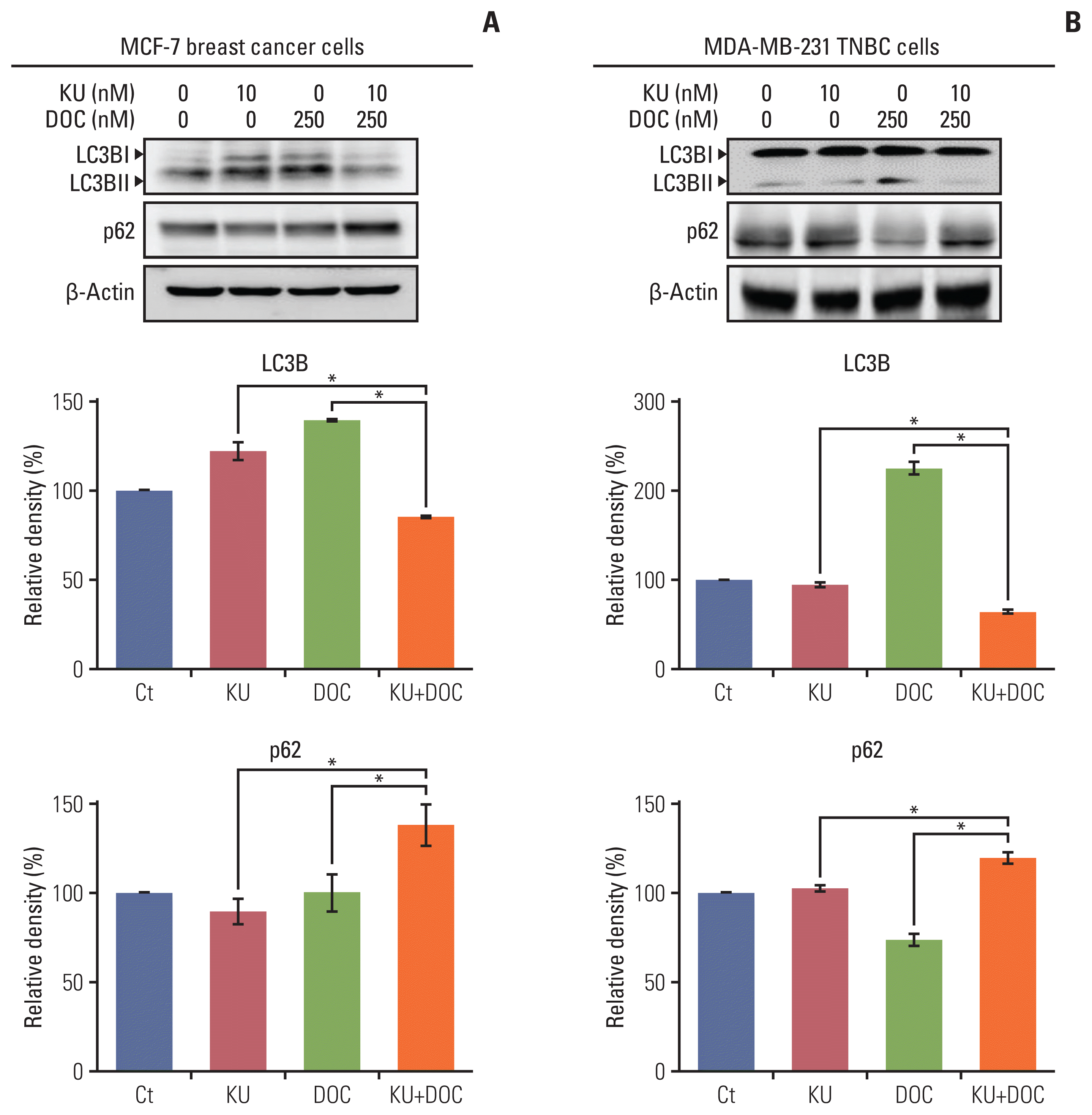 | Fig. 4Effects of docetaxel (DOC) and Ku-0063794 (KU), either individually or in combination, on the autophagy of breast cancer cells. (A) Western blot analysis showing the expression of autophagy-related markers in MCF-7 breast cancer cells following mono- and combination therapies of DOC and KU. Relative densities of individual markers. The relative densities had been quantified using Image J software and then were normalized to the density of β-actin in each group. (B) Western blot analysis showing the expression of autophagy-related markers in MDA-MB-231. TNBC cells following mono- and combination therapies of DOC and KU. Relative densities of individual markers. LC3B, microtubule-associated proteins 1A/1B light chain 3B. Values are presented as mean±standard deviation of three independent experiments. *p < 0.05.
|
5. Validation of combination therapy anticancer effects in vivo
Next investigation focused on the effects of docetaxel and Ku-0063794, treated either individually or in combination, on the growth of MCF-7 and MDA-MB-231 cells xenografted in nude mice. Xenograft models were generated by a 4-week intraperitoneal administration of MCF-7 and MDA-MB-231 cells (5×106 cells/mouse in 100 μL normal saline, three times a week). After the intraperitoneal administration of docetaxel (1 mg/kg/day) and Ku-0063794 (1 mg/kg/day) three times a week for 3 weeks, the mice were euthanized and the tumors were collected. Images of the tumors after necropsy showed that the shrinkage was most prominent in the mice treated with combination therapy than in the mice treated with individual monotherapies (Fig. 5A). In both types of breast cancer cells, a considerable reduction in tumor size was observed in the mice treated with combination therapy than in the mice with individual monotherapies (p < 0.05) (Fig. 5B). Collectively, the data presented here showed that combination therapy has a higher potential to reduce the growth of xenografted MCF-7 and MDA-MB-231 cells over individual monotherapies in the nude mouse model.
 | Fig. 5Effects of docetaxel (DOC) and Ku-0063794 (KU), individually and in combination, on the growth of MCF-7 and MDA-MB-231 cells xenografted into nude mice. After DOC (1 mg/kg/day) and KU (1 mg/kg/day) had been administered intraperitoneally three times a week for 3 weeks, the mice were euthanized, and the tumors were collected. (A) Morphological images of mice with xenografted MCF-7 and MDA-MB-231 cells following mono- and combination therapies of DOC and KU. Images of tumors after necropsy show that tumor shrinkage was more prominent in mice treated with combination therapy than in the mice treated with individual monotherapies. (B) Comparison of tumor size over time after injecting DOC and KU, either individually or in combination, into MCF-7 and MDA-MB-231 cells over time, respectively. In both types of breast cancer cells, a considerable reduction in tumor size was observed in mice treated with combination therapy than in mice treated with individual monotherapies. (C) Hematoxylin and eosin stains (top left) and cleaved caspase-3 (top middle) and Bcl-xL (top right) immunohistochemical stains of the MDA-MB-231 cells xenografted in nude mice after injecting docetaxel (DOC) and Ku-0063794 (KU), either individually or in combination. Percentages of cell count (bottom left) and immunoreactive areas (bottom middle and right) were measured using Image J and expressed as relative to the control. Values are presented as mean±standard deviation of three independent experiments. *p < 0.05.
|
6. Comparison of each group by tissue staining
Next, the xenografted MDA-MB-231 TNBC cells were stained to compare the effects of individual mono- and combination therapies (Fig. 5C). In hematoxylin and eosin staining, the number of tumor cells was significantly reduced after the combination therapy as compared to the individual monotherapy (p < 0.05) (Fig. 5C, left). Immunohistochemistry revealed that the expression of cleaved caspase-3 (a pro-apoptotic marker) was significantly higher in the mice with combination therapy than in those with individual monotherapies (p < 0.05) (Fig. 5C, middle). Furthermore, the expression of Bcl-xL (an anti-apoptotic marker) was significantly lower in the mice with combination therapy than in those with individual monotherapies (p < 0.05) (Fig. 5C, right). Taken altogether, these results suggest that the combination therapy has a higher potential of inducing apoptotic cell death than the individual monotherapies in MDA-MB-231 TNBC cells xenografted in the nude mice.
7. Validation of autophagy inhibition by combination therapy in vivo
The expression of autophagy markers, LC3B and p62, were determined in the MDA-MB-231 cells xenografted in the nude mice (Fig. 6A). Western blot revealed that the expression of LC3B was significantly increased in the docetaxel group but was not significantly changed in the Ku-0063794 and combination groups. In addition, the expression of p62 was decreased following individual monotherapies and significantly increased following combination therapy, suggesting that autophagy is inhibited after combination therapy. Immunofluorescence results confirmed the western blot analysis (Fig. 6B); combination therapy did not increase the expression of LC3B, but increased the expression of p62, suggesting the inhibitory effects on autophagy by combination therapy.
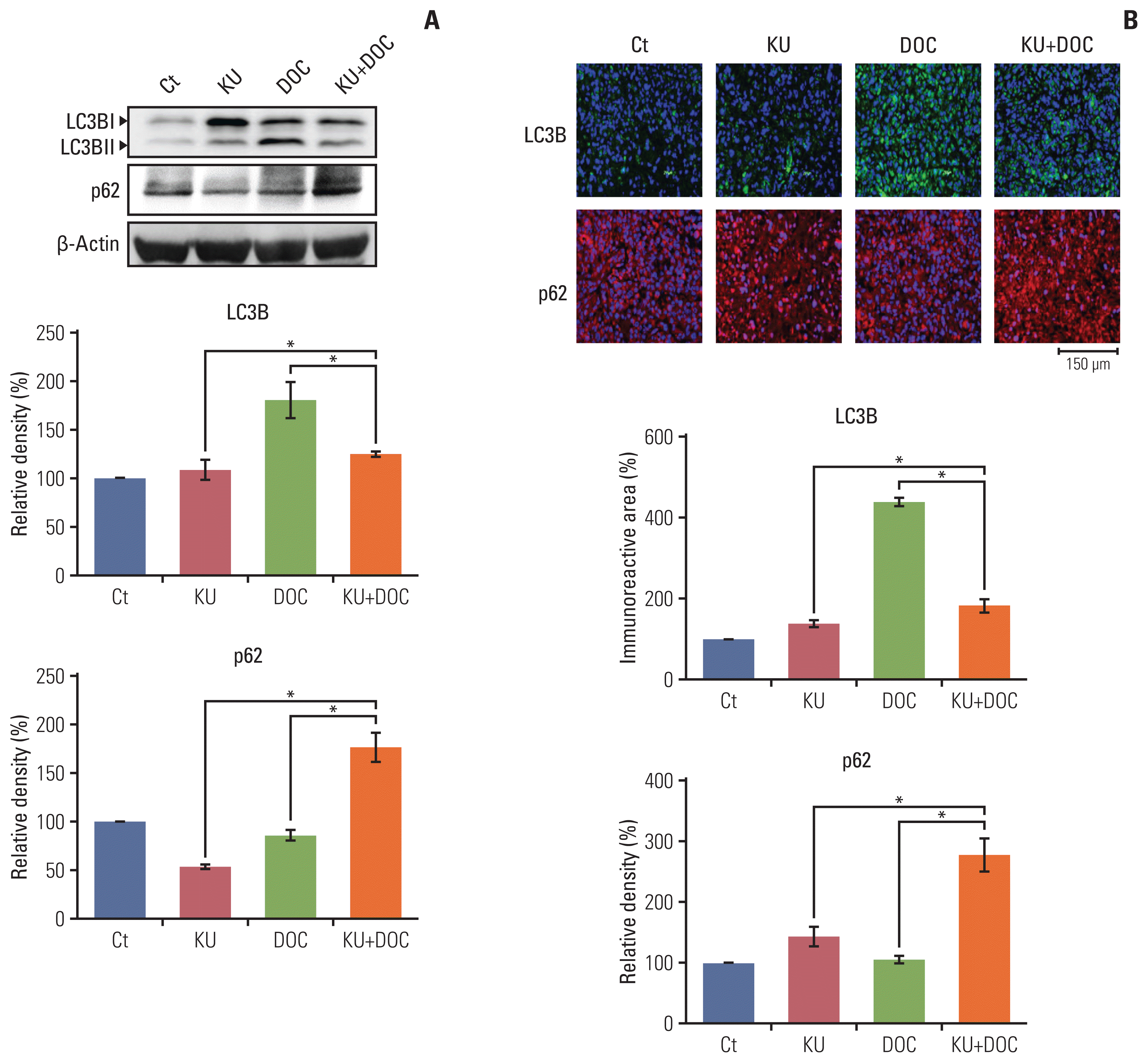 | Fig. 6Effects of each treatment on autophagy of triple-negative breast cancer (TNBC) cells in vivo. (A) Western blot analysis showing the expression of autophagy-related markers in MDA-MB-231 TNBC cells xenografted in nude mice following docetaxel (DOC) and Ku-0063794 (KU), mono- and combination therapies. Relative densities of LC3B and p62 proteins. The relative densities had been quantified using Image J software and then were normalized to the density of β-actin in each group. (B) LC3B and p62 immunofluorescence of MDA-MB-231 TNBC cells xenografted in nude mice following DOC and KU mono- and combination therapies. Percentages of immunoreactive areas were measured using NIH image J and expressed as relative values to the control. LC3B, microtubule-associated proteins 1A/1B light chain 3B. Values are presented as mean±standard deviation of three independent experiments. *p < 0.05.
|
8. Validation of the effects of combination therapy on EMT in vivo
Finally, western blot analysis was carried out to determine the effects of mono- and combination therapies on the expression of EMT-related markers in the MDA-MB-231 cells xenografted in the nude mice (Fig. 7A). In this experiment, increased epithelial maker (E-cadherin) and decreased mesenchymal markers (vimentin and snail) were considered to be the promotion of EMT. KU monotherapy slightly decreased EMT, considering the higher expression of E-cadherin and the lower expression of snail. By contrast, docetaxel monotherapy significantly increased EMT, considering the lower expression of E-cadherin and the higher expression of vimentin and snail (p < 0.05). And, the combination therapy led to the significant reduction of EMT, considering the higher expression of E-cadherin and the lower expression of vimentin and snail (p < 0.05). These results were also re-affirmed by the E-cadherin, vimentin, and snail immunofluorescences of the MDA-MB-231 cells xenografted in the nude mice (Fig. 7B).
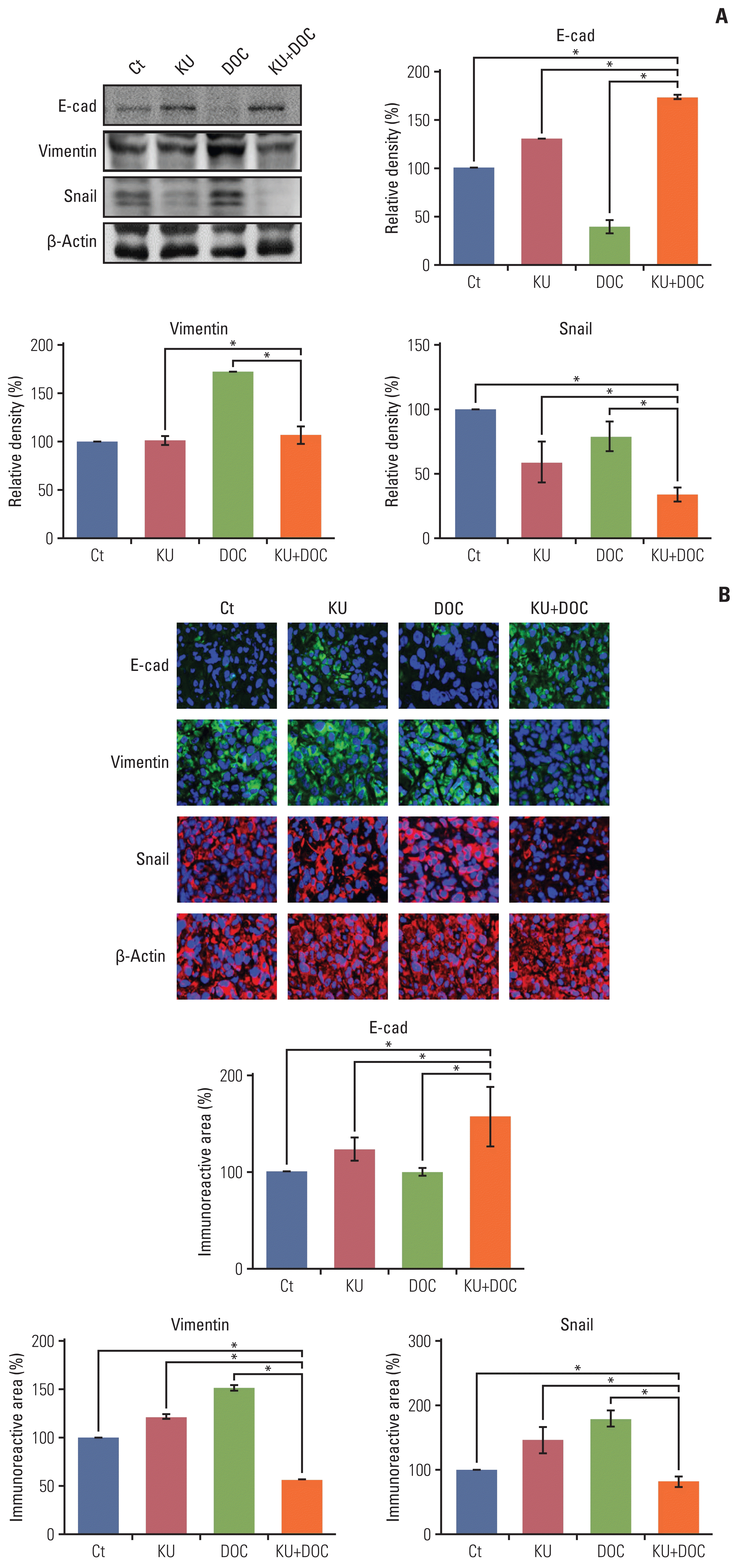 | Fig. 7Effects of each treatment on epithelial-mesenchymal transition (EMT) of triple-negative breast cancer (TNBC) cells in vivo. (A) Western blot analysis showing the expression of EMT-related markers in MDA-MB-231 TNBC cells xenografted in nude mice following docetaxel (DOC) and Ku-0063794 (KU) mono- and combination therapies. Relative densities of EMT-related markers. The relative densities had been quantified using Image J software and then were normalized to the density of β-actin in each group. (B) E-cadherin, snail, and vimentin immunofluorescence of MDA-MB-231 TNBC cells xenografted in nude mice following DOC and KU mono- and combination therapies. Percentages of immunoreactive areas were measured using NIH image J and expressed as relative values to the control. E-cad, E-cadherin; LC3B, microtubule-associated proteins 1A/1B light chain 3B. Values are presented as mean±standard deviation of three independent experiments. *p < 0.05.
|
Go to :
Discussion
To determine the therapeutic potential of docetaxel and Ku-0063794 combination therapy on the MDA-MB-231 TNBC cells, comparison between individual monotherapies and combination therapy was carried out with in vitro and in vivo experiments. In the in vitro experiments, the combination therapy was found to synergistically reduce the viability of MDA-MB-231 cells. Western blot analysis and flow cytometric analysis showed that the combination therapy induced higher apoptotic cell death than the individual monotherapies. Moreover, western blot analysis demonstrated that while each monotherapy increased the processes of EMT and autophagy, combination therapy decreased both. In the in vivo experiment, combination therapy was shown to have the higher potential to reduce the growth of xenografted MDA-MB-231 cells over individual monotherapies. In addition, the results of in vivo experiments were consistent with in vitro experiments, including the significantly higher inhibitory effects of the combination therapy on autophagy and EMT. Taken altogether, these data suggested that docetaxel and Ku-0063794 combination therapy had higher anticancer activities over individual monotherapy against MDA-MB-231 TNBC cells through the increased inhibition of autophagy and EMT processes.
mTOR complexes (mTORC1 and mTORC2) are essential mediators in the phosphoinositide 3-kinase, protein kinase B (Akt/PKB) and mTOR signaling pathway, and are crucial for cell growth, survival, motility, proliferation, protein synthesis, and transcription [12]. The first generation mTOR inhibitors that primarily target mTORC1 (such as temsirolimus and everolimus) are already FDA-approved for clinical use [12]. Unfortunately, the effectiveness of these inhibitors as a single agent therapy is stifled in part by strong mTORC1-dependent negative feedback loops that become inactive on mTORC1 inhibition [8–10,12]. In recent randomized phase II trial, the paclitaxel/cisplatin and everolimus combination was associated with more adverse events without improvement in pathological complete response or clinical response in patients with TNBC [13]. Therefore, the present study focused on the combined use of docetaxel (taxane-based regimen) and Ku-0063794 (a novel mTORC1/2 dual inhibitor) as new therapeutic agents for the treatment of TNBC. It inhibits Akt as well as the serum- and glucocorticoid-inducible protein kinases that are likely to play vital roles in driving the proliferation of many cancers [8,12,14,15]. The finding that Ku-0063794 induces more marked dephosphorylation of 4E-BP1 than rapamycin also holds promise that this drug would be more effective at suppressing protein synthesis required for growth and proliferation of cancer cells than rapamycin derivatives [8,15].
In this study, it was found that whereas individual monotherapies slightly decreased or rather increased EMT, combining both medications significantly reduced EMT of TNBC cells. EMT guides the transformation of non-mobile epithelial-like cells into mobile, mesenchymal-like cells, allowing tumor cells to acquire the capacity to infiltrate surrounding tissues and to metastasize to distant sites [16]. EMT is basically characterized by the loss of epithelial markers (downregulation of E-cadherin, occludins, claudins, desmoplakin, and epithelial cytokeratins) and the gain of mesenchymal (upregulation of vimentin, N-cadherin, fibronectin, and α-smooth muscle actin) markers expression in breast cancers [17,18]. EMT process is influenced by a variety of medications, including chemotherapeutic regimens. In our study, docetaxel showed a tendency to increase EMT in breast cancer cells. It has been noted that docetaxel could induce autophagy as well as apoptosis of cancer cells [19,20]. The promoted autophagy by docetaxel is implicated in the cancer cell resistance to chemotherapy, and thus could be related with an increase in EMT of cancer cells. In addition, the same medication could have different effects on the expression of EMT markers when co-administered with other medication(s). In our previous study, it was demonstrated that although everolimus and Ku-0063794 monotherapies did not significantly affect EMT in the hepatocellular carcinoma cells, combining both medications significantly reversed EMT process [21]. Similarly, in this study, it was found that although docetaxel and Ku-0063794 monotherapies could not inhibit EMT, combining both medications effectively inhibited EMT. We believe that the EMT-inhibiting ability of combination therapy could have contributed to the high synergistic effect of the combination therapy.
Autophagy is the major cellular catabolic degradation process in response to stressors such as nutrient deprivation and bioenergetic stress [22,23]. mTORC1 coordinates both anabolism and catabolism to meet the needs of cell growth. mTORC1 also inhibits autophagy induction, primarily by (1) inhibiting ULK1/2 and the VPS34 complex, and (2) by preventing global expression of lysosomal and autophagy-related genes through transcription factor EB phosphorylation [22–24]. In addition, it is generally accepted that mTORC2 also inhibits autophagy by activating mTORC1 through its interaction with ribosomes [24]. Thus, mTOR inhibitors basically have pro-autophagic properties. This experiment showed that Ku-0063794 had pro-autophagic effects in TNBC cells as demonstrated by up- and downregulation of LC3B and p62, respectively. Docetaxel is also known to have pro-autophagic properties [25]. Autophagy could be used by tumor cells as a way of escaping apoptotic cell death caused by chemotherapeutic regimens, thereby promoting chemo-resistance. Hu et al. [25] reported that docetaxel-mediated autophagy induced chemo-resistance in castration-resistant prostate cancer cells, and inhibiting autophagy led to the increased chemo-sensitivity.
Our results show that whereas Ku-0063794 and docetaxel monotherapies increased autophagy, combining both medications reduced autophagy. It occasionally occurs that combining several medications could lead to the alternations in the action of mechanism. This inhibition of autophagy was accomplished by down-regulating SIRT1. SIRT1 is an essential element of autophagic processes because it significantly contributes to autophagy by deacetylating essential autophagy-related proteins, such as Atg5, Atg7, and Atg8 [26]. While everolimus and Ku-0063794 monotherapies was found to increase SIRT1, combining both resulted in the reduced expression of SIRT1 [27]. Likewise, in our study, combining docetaxel and Ku-0063794 led to the inhibition of autophagy, of which mechanism needs to be further validated. We believe that reduced autophagy in combination therapy could have contributed to the synergistic effect of the anticancer effect in combination therapy.
In conclusion, this study showed that docetaxel and Ku-0063794 combination therapy has superior anticancer activities over individual monotherapy against MDA-MB-231 TNBC cells. Specifically, in the in vitro experiments using MDA-MB-231 cells, the combination therapy was found to synergistically reduce the cell viability and induced the higher pro-apoptotic cell death than individual monotherapies. Moreover, in the in vivo experiment, combination therapy was shown to have the higher potential to reduce the growth of xenografted MDA-MB-231 cells over the individual monotherapies. In addition, both in vitro and in vivo experiments consistently validated that, unlike individual monotherapies, docetaxel and Ku-0063794 combination therapy significantly inhibited the processes of EMT as well as autophagy. Taken altogether, these data suggested that docetaxel and Ku-0063794 combination therapy had higher anticancer activities than the individual monotherapies against MDA-MB-231 TNBC through the increased inhibition of autophagy and EMT processes.
Go to :
 XML Download
XML Download