

 PDF
PDF Citation
Citation Print
Print


Introduction
Radiotherapy (RT) has been widely utilized for curative or palliative care of patients with different types of cancer. Approximately 50% of patients with cancer require RT at least once during their courses of treatment, although the percentage may differ depending on medical infrastructure and availability of RT [1,2]. However, the degree and duration of response to RT varies widely and is largely dependent on the innate radiosensitivity of tumors and the delivered radiation dose [3,4]. Currently, radiosensitivity is known to be mostly dependent on the histology of the tumor [3–5]. Furthermore, it has been demonstrated that the intrinsic radio-sensitivity of cancer cells is a major determinant of tumor response [6]. Certain molecular determinants involving DNA damage repair, oxidative stress, and apoptosis may affect radiosensitivity in cancer cells [7].
DNA damage repair is necessary for the survival of both tumor cells and normal cells [8]. Damaged DNA is efficiently repaired via an intricate repair mechanism. For instance, ataxia-telangiectasia mutated (ATM) plays an important role in the repair of DNA double-strand breaks (DSBs). BRCA1 and 2 are the other important DSB repair proteins predominantly involved in the homologous recombination-repair (HR) pathway. It is well-documented that defects in DNA damage repair are associated with increased risk of toxicity and secondary malignancies after RT. For example, unexpected extreme radiation reaction was first reported in a 10-year-old boy with autosomal recessive ataxia-telangiectasia (A-T) disorder (caused by a germline mutation in ATM kinase) who underwent conventional 30 Gy of RT and died after 8 months [9]. Increased risk of contralateral breast cancer was reported in women with germline BRCA1/2 mutations who underwent lumpectomy and breast RT [10]. Increase in knowledge regarding unexpected radiation reaction has alerted clinicians when treating patients with the above genotypes with RT.
However, patients with somatic mutations in DNA repair genes in tumors, which occur more frequently than germline mutations, differ from patients with germline mutations, as the former do not harbor mutations in normal cells. Therefore, severe toxicities or secondary malignancies are not encountered frequently in these patients with cancer. Mutations in ATM and BRCA1/2 are common in various solid tumors [11,12]. Considering that the tumor cell killing effect of RT is mainly driven by lethal DNA damage, RT may also be considered as a critical component of treatment in patients harboring mutations in genes responsible for DNA damage repair. However, clinical data regarding whether tumors with ATM or BRCA1/2 somatic mutations are more sensitive to RT are scarce. Considering the increased use of multi-site palliation with advanced RT technologies, better understanding of the relationship between clinical RT response and such somatic mutations can facilitate the development of personalized RT regimens in the future.
In this study, we aimed to determine the effect of somatic ATM and BRCA1/2 mutations on radiosensitivity and treatment response. In this regard, we retrospectively analyzed the medical records of patients with solid tumors that received RT and compared the in-field target lesion control rates according to the presence of ATM and/or BRCA mutations. In addition, we compared the risk of RT-related toxicities between these two groups.
Materials and Methods
1. Study cohort
Between October 2013 and February 2019, 97 patients that received RT and underwent gene-panel sequencing of the biopsied tumor were subjected to analysis. Frameshift, missense, or nonsense mutations in ATM and/or BRCA1/2 were detected in 33 patients (mutation group); six patients harbored mutations in both ATM and BRCA (ATMmtBRCAmt), five patients harbored mutant ATM but wild type BRCA (ATMmtBRCAwt), and 22 patients harbored BRCA mutation but wild type ATM (ATMwtBRCAmt). As a control cohort, 33 patients were 1:1 propensity score-matched to the mutation group. Matching was performed using the variables that may maximally affect the RT response, which included age, sex, radiosensitivity, and RT dose (equivalent dose in 2 Gy [EQD2], < 45 Gy vs. ≥ 45 Gy). Radiosensitivity was subjectively defined as either radioresistant (colorectal cancer and melanoma) or radiosensitive (breast, lung, and gynecological malignancy) according to previous reports and our clinical experiences [4,13–15].
2. Radiotherapy
All patients underwent a computed tomography (CT) simulation using an appropriate immobilization device if required. The gross tumor volume was defined as the visible tumor in CT or other imaging studies such as positron emission tomography (PET) or magnetic resonance imaging (MRI). Three-dimensional conformal RT or intensity-modulated RT was applied based on the tumor location and the physician’s discretion. The dose constraints for the normal organ were set according to the institutional consensus protocol and guideline of the Quantitative Analyses of Normal Tissue Effects in the Clinic (QUANTEC).
3. Follow-up and response evaluation
Initial follow-up for the patient was usually made one month after completion of RT. Patients were followed with 3-month interval during the first year and 6 months thereafter. Physical examination, basic laboratory tests, and imaging evaluations, including CT, MRI, or PET were performed during the follow-up. In-field tumor response was assessed according to Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 by a board certified radiologist (blinded read), and classified as complete response (CR), partial response (PR), stable disease, and progression of disease (PD). For patients who underwent follow-up PET, Positron Emission Tomography Response Criteria in Solid Tumors (PERCIST) was adopted to evaluate tumor response. Objective response was defined as either CR or PR. The objective response rate was defined as the proportion of patients achieving an objective response. For bone lesions, lytic or mixed lytic-blastic lesions with identifiable soft tissue components that were considered measurable were evaluated. The primary endpoints were best response to RT and the local recurrence rate (LRR). The second endpoint was toxicity. LRR was defined as the time from RT initiation to local recurrence or last follow-up. Local recurrence was defined as the PD at the irradiated site.
4. Next generation sequencing
Targeted DNA or RNA sequencing was performed using TruSight Tumor 170 (Illumina, San Diego, CA), which detects 170 cancer-related genes [16]. We have focused on genes related to DNA damage response pathway, such as ATM, BRCA1, BRCA2, TP53, and PALB2. Briefly, 40 ng DNA and RNA were extracted from formalin-fixed paraffin-embedded (FFPE) tissue using the Qiagen All Prep DNA/RNA FFPE kit (Qiagen, Hilden, Germany). After hybridization capture-based target enrichment, pairended sequencing (2×150 bp) was performed using a NextSeq sequencer (Illumina) according to the manufacturer’s instructions. Variant calling was performed using Illumina App-pipeline (Illumina). Variants with a total depth of at least 100× and variant allele frequency of at least 3% were included for analysis. Benign single nucleotide polymorphisms (≥ 1% frequency based population-based database) were filtered out. Variant interpretation was based on recommendations from the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists [17]. The clinical significance of mutations was classified by a four-tiered system; tier I, variants with strong clinical significance; tier II, variants with potential clinical significance; tier III, variants of unknown clinical significance; and tier IV, variants deemed benign or likely benign (S1 Table). Tier I, II, and III variants were included in our study.
5. Statistical analysis
Patient characteristics were compared using the chi-square test, Fisher exact test, Student’s t test, or the analysis of variance (ANOVA) as applicable. Cumulative LRR was calculated by competing risk analysis, and comparisons were made using Gray’s test. A competing event was defined as death from any cause without evidence of local recurrence. Propensity score matching was performed using MatchIt package ver. 3.0.2. A generalized linear model was used without a pre-specified caliper. Different variables, such as age, sex, radio-sensitivity, pre-RT chemotherapy, radiation dose (EQD2), and type of RT modality, were included for matching. Primary and irradiated sites were excluded since they were highly diverse compared to the number of patients matched. Logistic regression was performed to identify the significant predictors of objective response. Variables with p < 0.1 in the univariate analysis were included in the multivariate analysis. Two-sided p < 0.05 was considered statistically significant. Statistical analyses were conducted using IBM SPSS Statistics ver. 24.0 (IBM Corp., Armonk, NY), GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA), and R ver. 3.6.0 (https://www.r-project.org).
Results
1. Patient characteristics
Baseline characteristics of patients in the mutation group and wild type group before and after propensity score matching are shown in S2 Table. The median RT dose (EQD2) delivered was 44.7 Gy (range, 11.5 to 125 Gy) and it was dichotomized to < 45 Gy and ≥ 45 Gy. The characteristics were well balanced after matching (S2 Table). Patients in the mutation group were further stratified by ATM and BRCA mutation status (S3 Table). No significant differences in baseline characteristics were observed between the four groups: ATMwtBRCAwt, ATMmtBRCAwt, ATMwtBRCAmt, and ATMmt BRCAmt.
In total, 91 lesions were treated in the matched 66 patients where 46 lesions had neither ATM or BRCA mutations and 45 lesions harbored ATM and/or BRCA mutations. Nineteen patients had multiple lesions treated and 47 patients had a single lesion treated with RT. Clinical significance of mutations detected in ATM, BRCA1, and BRCA2 are summarized in S1 Table. None of the mutations were confirmed to be benign and 47.4%, 63.6%, and 41.9% of mutations were suggested to have at least potential clinical significance in ATM, BRCA1, and BRCA2, respectively. No significant differences in baseline characteristics were observed except for age, primary sites, and RT dose among the ATMwtBRCAwt, ATMmtBRCAwt, ATMwtBRCAmt, and ATMmtBRCAmt groups (Table 1). RT was delivered in diverse dose schemes, namely conventionally fractionated RT (21 lesions, 23.1%), hypofractionated RT (62 lesions, 68.1%), and stereotactic body radiation therapy (8 lesions, 8.8%). No significant differences in the RT dose scheme were observed among the four groups (Table 1). Most patients received RT to their metastatic lesions. Among the 91 lesions treated, 14 (15.4%) were matched with the sequenced tumor and 38 (41.8%) originated from the metastatic site (Table 1). The frequency of matched RT sites to biopsied sites and the sites of next generation sequencing performed did not significantly differ among the four groups.
2. Treatment outcomes
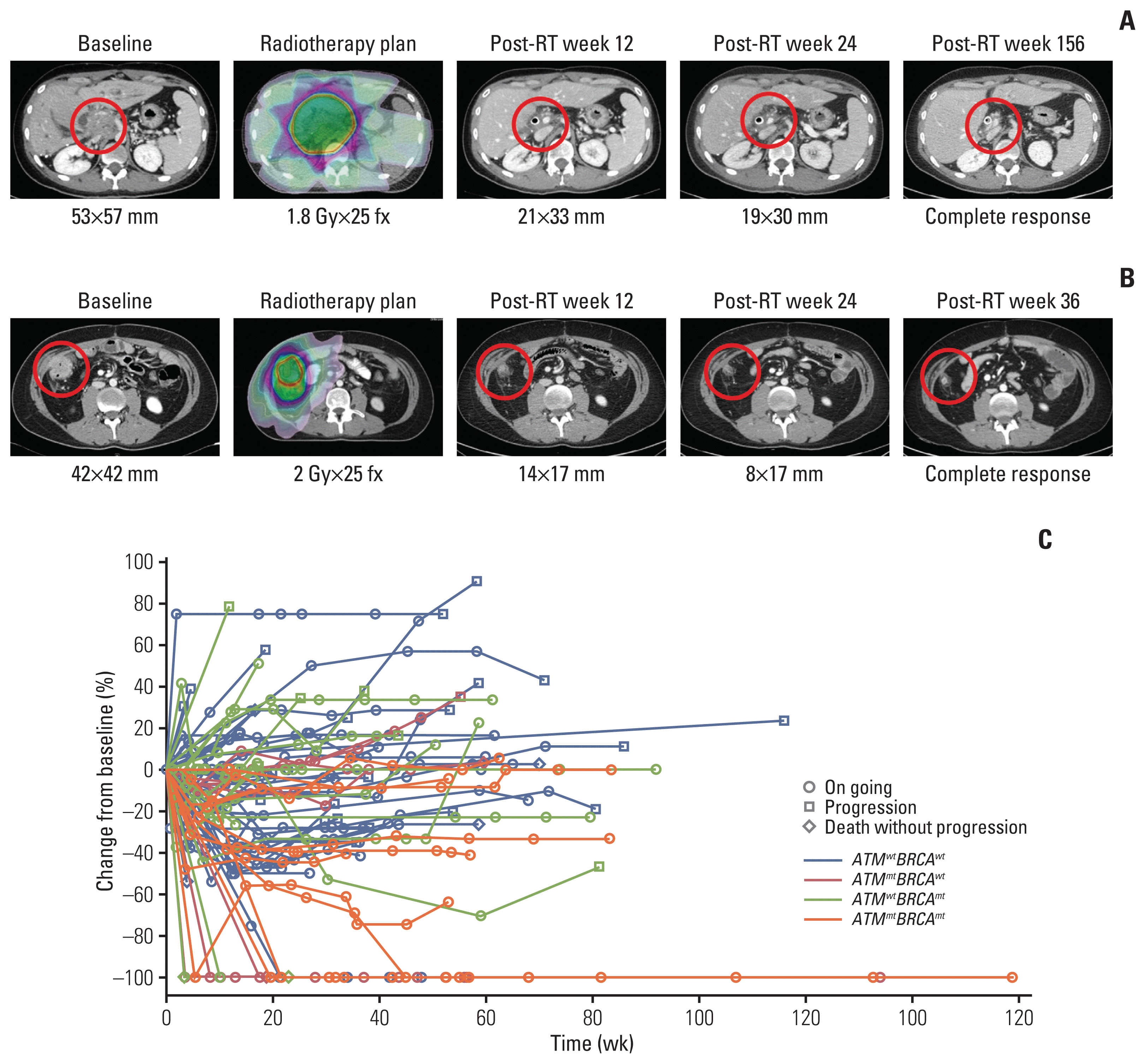
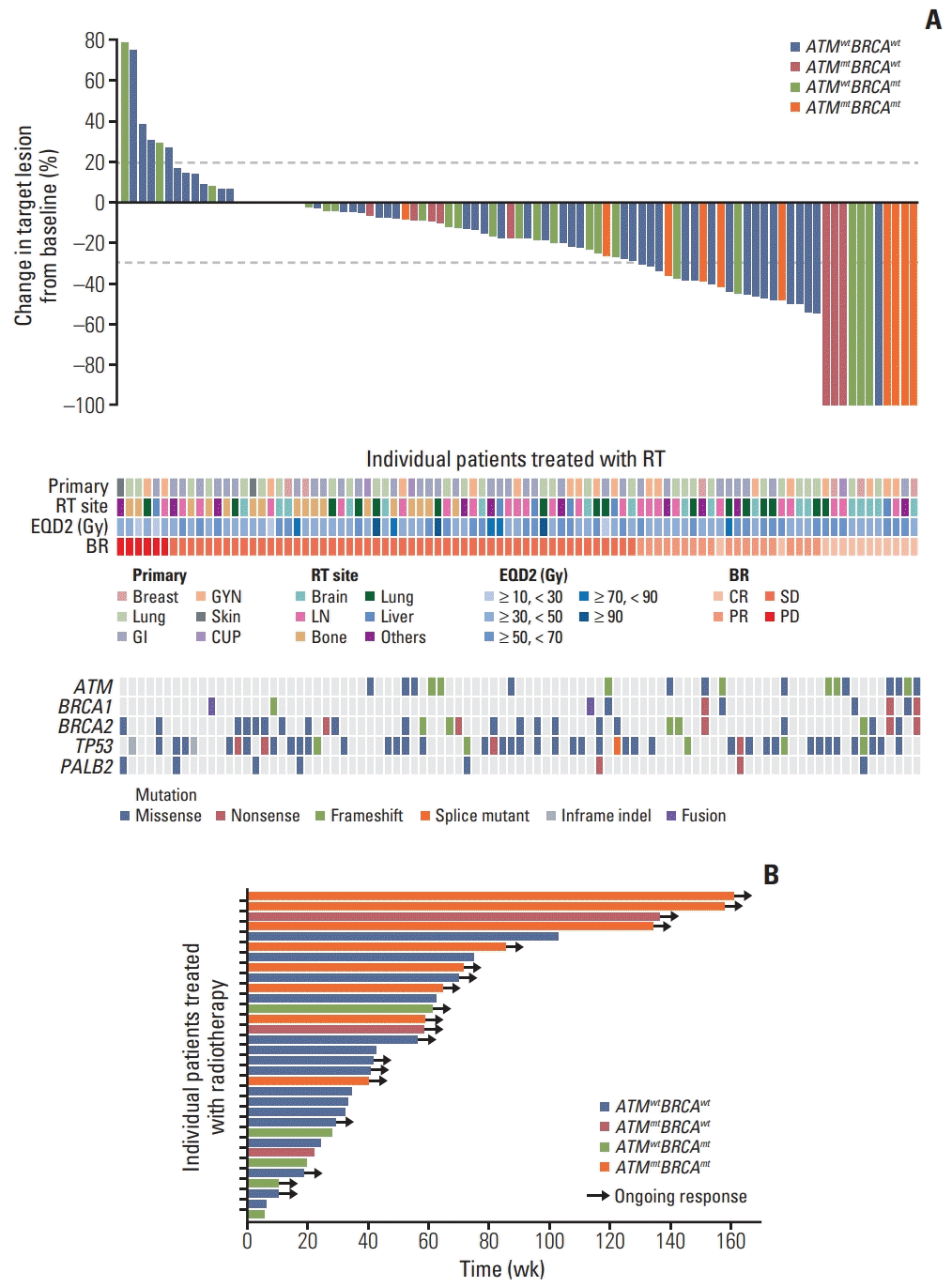
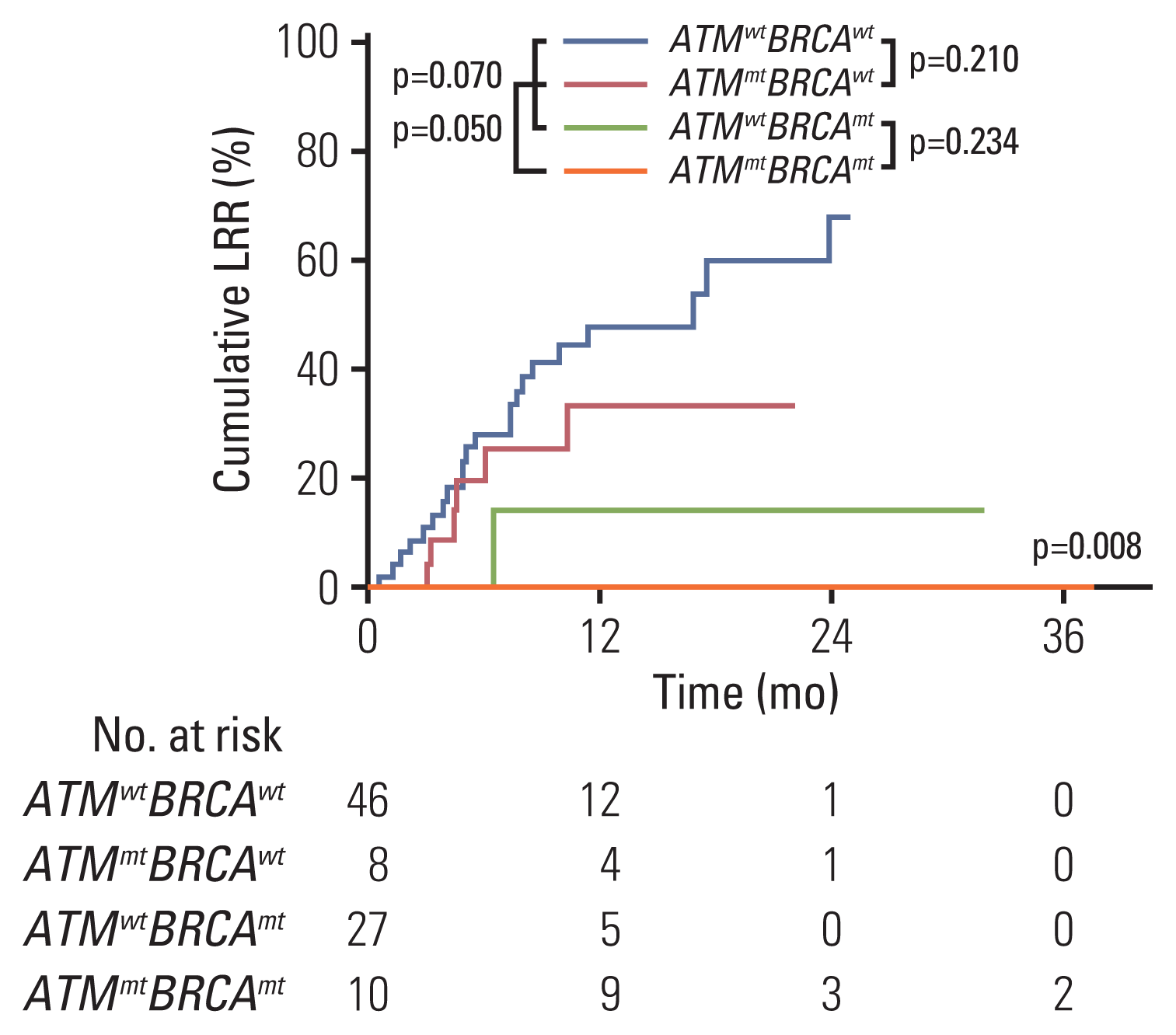
The median follow-up time was 13.9 months (range, 2.5 to 69.5 months). The treatment response following RT differed significantly among ATMwtBRCAwt, ATMmtBRCAwt, ATMwtBRCAmt, and ATMmtBRCAmt groups (Fig. 1). A representative ATMmtBRCAmt case that exhibited CR after RT and the serial change in the diameter of the longest tumor of all lesions are shown in Fig. 1. A majority of patients exhibited reduction in tumor size after RT. Objective response rates were 34.8%, 37.5%, 18.5%, and 80.0% in ATMwtBRCAwt, ATMmtBRCAwt, ATMwtBRCAmt, and ATMmtBRCAmt, respectively (p=0.007) (S4 Table). Notably, the highest CR rate of 60% was observed in the ATMmtBRCAmt group, whereas the ATMwtBRCAwt group showed the lowest CR rate of 2.2% (p < 0.001) (S5 Table). Two lesions with radiological PR and metabolic CR were considered to exhibit CR. Among the 19 patients that had multiple sites treated, 13 (68.4%) exhibited a mixed best tumor response, while six (31.6%) exhibited the same best tumor response. The profound treatment response in the ATMmtBRCAmt group is also shown in the waterfall plot (Fig. 2A). None of the lesions in the ATMmtBRCAwt group progressed after RT (Fig. 2A). Mutations in other genes related to DNA damage response pathway, such as TP53 and PALB2, are shown in Fig. 2A, although they did not correlate with response to RT. The median duration of response in patients that had an objective response were 8.7 months (range, 1.4 to 23.9 months), 13.6 months (range, 5.1 to 31.8 months), 4.5 months (range, 1.3 to 14.2 months), and 18.2 months (range, 9.3 to 37.5 months) in the ATMwtBRCAwt, ATMmtBRCAwt, ATMwtBRCAmt, and ATMmtBRCAmt groups, respectively (Fig. 2B). In addition, all eight lesions (100%) that exhibited an objective response in the ATMmtBRCAmt group exhibited an ongoing treatment response, while seven out of 16 lesions in the ATMwtBRCAwt group (43.8%), two out of three lesions in the ATMmtBRCAwt group (67%), and two out of five lesions in the ATMwtBRCAmt group (40%) exhibited ongoing response (Fig. 2B). In the univariate analysis, mutation group (p=0.023) was only significantly associated with objective response. Its significance was maintained in the multivariate analysis (p=0.016) (Table 2). The cumulative 1-year LRR in the ATMwtBRCAwt, ATMmtBRCAwt, ATMwtBRCAmt, and ATMmtBRCAmt groups were 47.9%, 14.3%, 33.2%, and 0%, respectively (p=0.008) (Fig. 3, S4 Table). The differences in cumulative LRR between the ATMwtBRCAwt, ATMmtBRCAwt, ATMwtBRCAmt, and ATMmtBRCAmt groups tend to be more prominent according to ATM mutation status rather than BRCA mutation status (Fig. 3).
3. Toxicity
The incidence of any treatment-related toxicities after RT was 19.6%, 12.5%, 11.1%, and 20.0%, in the ATMwtBRCAwt, ATMmtBRCAwt, ATMwtBRCAmt, and ATMmtBRCAmt groups, respectively (p=0.792). Most toxicities were limited to grade 1 or 2, and only one event of grade 3 nausea was reported (Table 3). Nausea was most common and occurred in treatments including brain or intra-abdominal lesions. Esophagitis and pneumonitis were reported in treatments including mediastinal lesions.
Discussion
In the present study, we found that tumors harboring mutations in genes related to DNA damage repair exhibit a superior treatment response to RT. Although most previous studies have focused on germline mutations [18,19], we utilized tumor tissues to account for the role of somatic mutations in these tumors. Another strength of this study is that we compared the outcome of RT in patients with mutations to propensity score-matched patients without any mutation. Response to RT varied among patients and depended on factors such as tumor histology, radiation dose, hypoxia, and intrinsic radiosensitivity of tumor cells [3–7]. Among multiple factors, intrinsic cancer cell radiosensitivity is the major factor determining RT response. Considering the mode of action of RT, which induces cell death due to generation of DNA DSBs, the ability of cancer cells to repair DNA damage can considerably affect their radiosensitivity [20].
DNA damage repair involves diverse pathways according to the type of damage induced [8]. DNA DSBs, which are the most common type of DNA damage induced by RT, are repaired via two distinct pathways for repair: the non-homologous end joining (NHEJ) pathway and the HR pathway The pathway that is activated to repair DNA DSBs depends on the cell cycle phase. NHEJ is dominant in the G1 phase and HR is dominant in the mid-S and mid-G2 phases [21]. Although NHEJ is known to be the major pathway activated to repair DNA DSBs in mammalian cells, HR may also have role in DNA DSB repair [8]. Both BRCA and ATM play crucial roles in the repair of DNA DSBs [22,23]. Germline mutation in ATM can cause A-T syndrome that is characterized by increased sensitivity to RT [9]. BRCA1/2 mutation carriers with breast cancer have a higher risk of contralateral breast cancer after breast RT than controls with sporadic breast cancer [10].
BRCA1 and 2 are key molecules mediating the HR pathway, and BRCA mutations can disrupt DNA DSB repair [24]. In contrast, compared to BRCA, ATM is involved in the earlier steps of DNA damage repair, as it acts as a sensor of DNA damage [25]. Preclinical studies have demonstrated that pharmacological inhibition of ATM function enhances radiosensitivity [26]. Previously, a case series reported exceptional response to RT in a clinical setting involving eight patients with somatic mutations in ATM [27]. We also observed that patients with ATM or BRCA mutations show better treatment response and lower LRR. Furthermore, patients with ATM mutations tended to show better response to RT than those with BRCA mutations. ATM not only plays a crucial role in HR but is also involved in NHEJ, the major pathway for DNA DSB repair [28]. As BRCA is mostly involved in HR alone, we suggest that the impact of ATM mutations may be more pronounced than BRCA mutations in DNA repair dysfunction. TP53 and PALB2, the DNA damage response pathway-related genes included in the next generation sequencing (NGS) panel, did not show clear association with treatment outcome. A previous report also demonstrated lack of association between TP53 mutation and response to RT [29]. Considering the dominant role of NHEJ in DNA DSB repair, the correlation of somatic alterations in NHEJ pathway genes and response to RT should be investigated in future studies. However, a recent study that analyzed the frequency of somatic alteration in DNA damage repair genes in the Cancer Genome Atlas across 33 cancer types and demonstrated that more HR pathway genes than NHEJ pathway genes were included in the 50 most frequently mutated DNA damage repair genes [30]. ATM was the most commonly mutated gene among DNA DSB repair genes. Due to the lower frequency of somatic mutations in NHEJ pathway genes than HR pathway genes, a larger cohort should be analyzed to properly address the impact of somatic mutations in NHEJ pathway genes on responsiveness to RT.
We also observed that tumors with both ATM and BRCA mutations have an exceptional response to RT, which was durable. Surprisingly, no local recurrence occurred in lesions with both ATM and BRCA mutations. Loss-of-function of either ATM or BRCA alone can lead to activation of alternative pathways to compensate for the dysfunctional DNA damage repair process [21]. However, loss-of-function of both ATM and BRCA may lead to blockade of both HR and NHEJ pathways, which leads to a more pronounced defect in DNA DSB repair. Considering the exceptional response of ATMmt BRCAmt tumors to RT with a median dose of 45 Gy, we speculated that these tumors may respond well to even lower doses of RT. A previous study demonstrated that ATM-deficient cells show defects in DNA DSB repair in an RT dose-independent manner, and that DNA damage persisted even after 0.02 Gy of irradiation [31]. These data indicated the necessity of investigating dose de-escalation while treating ATMmtBRCAmt tumors.
One of the concerns of treating patients with mutations in DNA damage repair-related genes is the higher risk of developing toxicity and secondary malignancy [19,32]. The chances of developing toxicity and secondary malignancy are low as long as the mutation is not present in the germline, as normal cells do not harbor such mutations and have intact DNA damage repair pathways. In our study, we did not observe any significant increase in toxicity after irradiating tumors with ATM or BRCA mutations, which implies that RT dose constraints similar to those used for normal organs may be used in these patients compared to those without mutations.
This study had several limitations which made the results less conclusive. The sample size was small due to the limited number of patients who received both RT and underwent NGS. Characteristics of the patients were also heterogeneous regarding RT dose and primary site. In addition, we were only able to match patients with or without mutations rather than the four subgroups due to the limitation in the number of patients, which resulted in significant differences in age and primary site among the subgroups. Furthermore, we were not able to analyze the effect of mutations according to tumor histology or different RT doses. Further analysis in a larger cohort of patients is required to confirm our results. The follow-up period was relatively short to account for late RT-related toxicities and secondary malignancies. Furthermore, sequencing was only performed in tumor tissue and we were not able to properly filter germline variants. However, variant allele frequency (S6 Fig.) suggested that most mutations are possibly somatic instead of germline. Among DNA damage repair genes, mutations in only ATM and BRCA were evaluated, as other HR and NHEJ pathway-related genes were not included in the NGS panel. For competing risk analysis of multiple lesions treated in the same individual, a stratified analysis that adjusts for the effect of each individual should be performed. However, in the present study, this could not be performed due to the lack of local recurrence events in the ATMmtBRCAmt group [33]. Moreover, multivariate analysis of LRR could not be performed due to the lack of local recurrence events in the ATMmtBRCAmt group.
In this hypothesis generating study, we found that tumors with ATM or BRCA mutations may have enhanced radiosensitivity, and that the presence of both mutations may lead to exceptional response to RT. In the era of personalized medicine, continuous efforts are being made to customize RT dose according to the intrinsic radiosensitivity of the tumor [7,34]. Taking advantage of NGS, we can now easily evaluate the patients’ mutation profile to identify radiosensitizing mutations, such as in ATM and BRCA [35]. Further investigations are required to confirm whether such mutations confer radiosensitivity and whether de-escalation of RT dose is feasible for tumors harboring such mutations. In addition, future studies can focus on using combinations of DNA repair inhibitors. As DNA repair inhibitors have entered clinical trials as single agents and as image-guided, intensity-modulated radiation therapy has been successful in decreasing RT toxicity, the onus lies on the radiation oncology community to carefully design clinical trials.
 XML Download
XML Download