

 PDF
PDF Citation
Citation Print
Print


Introduction
Atypical teratoid/rhabdoid tumor (ATRT) of the central nervous system (CNS) is a highly aggressive malignancy, accounting for 1%–2% of all pediatric CNS tumors [1,2]. Among children under the age of 3 years, ATRT constitutes the most common malignant tumor of CNS (17.3%), followed by medulloblastoma (16%) [3].
Since standard therapeutic strategies for ATRT have not been established yet, treatment approaches for ATRT vary among institutions and countries. Conventional chemotherapy in addition to high-dose chemotherapy (HDCT) with autologous stem cell rescue (ASR) is usually utilized in pati-ents under 3 years of age to substitute for or delay the use of radiotherapy (RT) in attempt to improve outcomes and minimize long-term neurocognitive impairment [4]. However, most patients with ATRT suffer rapid disease progression (PD), although treatment regimens designed for CNS neoplasms have been used [5,6]. Outcome for patients with ATRT is generally dismal, particularly in the presence of residual tumor or metastasis. ATRT is a devastating brain neoplasm with a median survival time ranging from 6 to 13 months. This is caused not only by the disease’s aggressive characteristics, but also by a lower tolerance of young patients to treatment. This is exacerbated by the hesitancy to use RT in younger patients due to risks of functional impairment of the developing brain [7]. It has been shown that aggressive therapy can prolong survival in a subset of children with CNS ATRT [1,2,8]. Some clinical trials have incorporated early administration of RT to the primary site together with intrathecal (IT) chemotherapy, resulting in improved outcomes [9,10].
In 2005, the Korean Society for Pediatric Neuro-Oncology (KSPNO) suggested the following a protocol for ATRT patients aged less than 3 years (KSPNO-S052): six cycles of conventional chemotherapy and tandem HDCT/ASR are performed initially and RT is deferred until the patient reaches 3 years of age. In 2008, a minor revision of the protocol including a dose modification of conventional chemotherapy was made (KSPNO-S082). In 2011, the protocol was revised (KSPNO-S1102) to recommended early local RT with concurrent chemotherapy within four weeks after surgery. In addition, IT chemotherapy was incorporated.
Given the rarity of this tumor and the even lower number of very young infants affected by ATRT, no detailed clinical analyses have been directed toward patients under 3 years of age. Thus, the objective of this study was to describe patients diagnosed with ATRT in this age group and determine potential specific characteristics or prognostic factors. Their therapeutic management is also suggested.
Go to : 
Materials and Methods
1. Data collection
Children younger than 3 years of age at diagnosis who were newly diagnosed with CNS ATRT were eligible for this study. A search of medical records from seven centers was performed to identify patients aged 3 years or less who were diagnosed between January 2005 and December 2016. In cases with multiple intracranial and extracranial rhabdoid tumors, only patients with clear descriptions of a primary tumor within the CNS were included in this analysis. All participating centers received Institutional Review Board approval to contribute data for this study.
2. Staging
Proper staging for metastases included brain and spinal magnetic resonance imaging (MRI) and cytology of the cerebrospinal fluid (CSF). Modified Chang status for metastatic stage was recorded whenever available. It was defined as follows: M0, absence of metastases; M1, presence of metastases confined to CSF; M2, presence of metastases in the brain; M3, presence of metastases in the spinal subarachnoidal space; and M4, spread outside of the CNS [11]. M+ included M1, M2, M3, M4, and any metastasis not further described.
3. Treatment
All patients underwent maximal possible surgical resection of the primary lesion to preserve neurologic function. The extent of surgical resection defined as gross total resection (GTR), subtotal resection, or biopsy was determined based on a review of postoperative MRI and the surgeon’s intraoperative assessment. Multimodal therapies including surgery, RT, chemotherapy and HDCT/ASR were performed. Treatment was mainly performed according to KSPNO recommendations. However, some physicians modified the treatment protocol depending on clinical situation. Treatment scheme of the KSPNO regimen is shown in Fig. 1. Major differences between KSPNO-S052/-S082 (pre-2011) and KSPNO-S1101 (post-2011) were the timing of RT (delayed RT pre-2011 vs. early adjuvant RT post-2011) and IT chemotherapy for patients post-2011. Detailed chemotherapy schedules are described in Table 1.
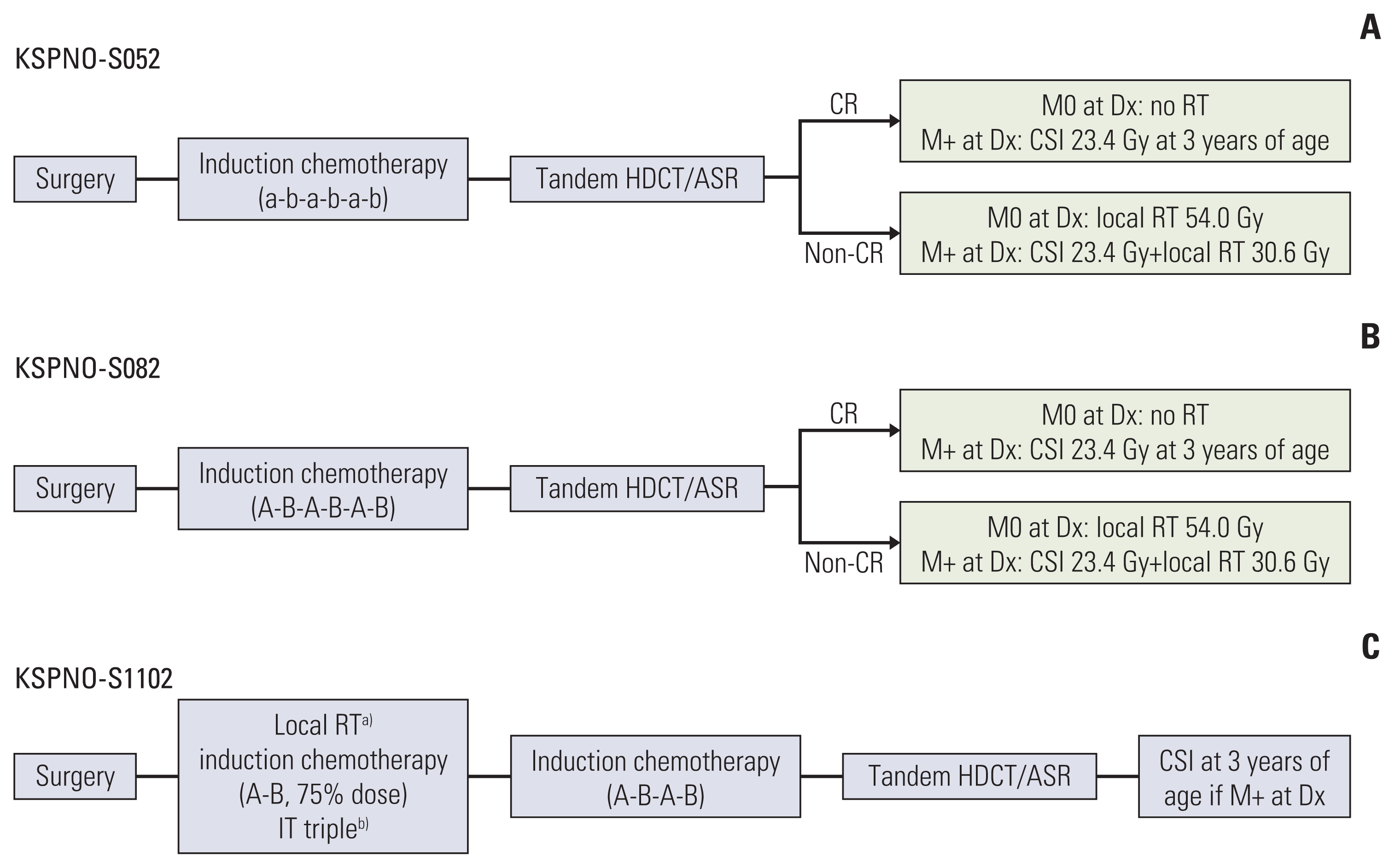 | Fig. 1Treatment recommendations by the Korean Society for Pediatric Neuro-Oncology for CNS ATRT during periods of 2005–2007 (A), 2008–2010 (B), and 2011-present (C). ASR, autologous stem cell rescue; CR, complete response; CSI, craniospinal irradiation; HDCT, high-dose chemotherapy; IT, intrathecal; RT, radiotherapy. a)R0 (< 1.5 cm2)/M0, local RT 41.4 Gy; R+ (> 1.5 cm2)/M0, local RT 55.8 Gy; Rx/M+, local RT 36 Gy+CSI 19.8 Gy at 3 years of age, b)M0, day 0 of each cycle/M+, weekly until clearing (at least 6 times), then day 0 each cycle.
|
Table 1
Chemotherapy regimens
![]()
KSPNO-S052/S082 (pre-2011) suggested the following: Induction treatment was initiated within 4 weeks of surgery. Six cycles of chemotherapy using alternating CECV (cisplatin, etoposide, cyclophosphamide, and vincristine) and CEIV (carboplatin, etoposide, ifosfamide, and vincristine) regimens were administered. Additionally, tandem HDCT/ASR was performed. The first course included carboplatin, thiotepa and etoposide (CTE), and the second course included cyclophosphamide and melphalan (CM). A 12- to 16-week interval between the first and second HDCT/ASR was allowed to minimize treatment-related mortality (TRM). RT was deferred until after 3 years of age unless the tumor showed progression or relapse. For patients with M0 disease at diagnosis and for patients those without residual tumor after HDCT, RT was omitted.
KSPNO-S1102 (post-2011) suggested the following: RT was recommended to be administered within 4 weeks of surgery with concurrent chemotherapy. IT chemotherapy including cytarabine, hydrocortisone, and methotrexate (MTX) was administered on day 0 of every chemotherapy cycle. For M+ patients, weekly IT chemotherapy was administered until clearance of CSF cytology. After six cycles of alternating CECV and CEIV regimens, tandem HDCT/ASR using CTE-CM was performed. For M+ patients at diagnosis, craniospinal irradiation (CSI) was deferred at 3 years of age.
4. Response and toxicity criteria
Disease response was evaluated by MRI and CSF cytology. Evaluations were repeated every two or three chemotherapy cycles prior to the first HDCT/ASR, between the first and second HDCT/ASR, every 3 months for the first year after completion of tandem HDCT/ASR, every 4 months for the second year, and every 6 months thereafter. Disease response was categorized as follows: complete response (CR) for complete disappearance of all tumors, partial response (PR) for decrease in tumor size by more than 50%, stable disease for less than 25% change in tumor size, PD for greater than 25% increase in tumor size or the appearance of new tumors. Toxicities were graded using the National Cancer Institute’s Common Terminology Criteria ver. 4.0.
5. Statistical analysis
The cutoff point for data analyses was March 2019. For descriptive statistics, data were compared using the Fisher exact test for categorical factors and Wilcoxon Mann-Whitney U test for continuous factors. The time to progression was calculated from the date of diagnosis until the date of PD. Progression-free survival (PFS) was calculated from the date of diagnosis to the date of PD or relapse. Overall survival (OS) was calculated from the date of initial diagnosis to the date of last follow-up or death from any cause. PFS and OS were estimated using the Kaplan-Meier method. Univariate analysis of risk factors was performed by comparing PFS and OS using the log-rank test. Multivariate logistic regression was used to examine relationships between outcomes of PFS or OS as binary dependent variables and independent variables of patient age, metastases, extent of resection, early adjuvant RT, HDCT, and years of diagnosis. p < 0.05 was considered statistically significant. All analyses were performed using IBM SPSS ver. 18.0 (SPSS Inc., Chicago, IL).
Go to :
Results
1. Patient characteristics
Baseline clinical characteristics of patients are shown in Table 2. Forty-three patients were enrolled in this study. The median age at diagnosis was 13 months (range, 0 to 32 months). Twenty patients (46.5%) were younger than 1 year of age at diagnosis. One patient presented with synchronous tumors, including ATRT in brain and a rhabdoid tumor in the kidney.
Table 2
Clinical characteristics of patients
![]()
Nineteen patients (44.2%) were diagnosed before 2011 (pre-2011 group) and 24 patients (55.8%) were diagnosed after 2011 (post-2011 group). There was no significant difference in age at diagnosis (p=0.55), sex (p=0.43), extent of resection (p=0.15), or metastases (p=0.46) between pre-2011 and post-2011 groups. There were 23 (53.5%) patients with M0 disease, four (9.3%) with M1 disease, 14 (32.6%) with M2 disease, and two (4.7%) with unknown metastatic status. The incidence of metastatic disease was significantly higher in patients under 6 months of age than that in patients older than 6 months (100.0% vs. 35.3%, p < 0.01). GTR of the primary tumor was achieved in 24 patients (55.8%).
2. Treatment
One patient did not receive any further therapy after surgery. All remaining 42 patients received induction chemotherapy at a median of 20 days after surgery (range, 5 to 142 days). The median number of pretransplant chemotherapy cycles was 6 (range, 1 to 12). All patients in the post-2011 group received IT therapies. Nine patients (21.4%) received a second surgery before HDCT due to PD (n=8) or for removing residual tumor (n=1).
Twenty-nine patients (69.0%) received radiation at a median age of 23 months (range, 6 to 40 months). The median interval between diagnosis and RT was 162 days (range, 21 to 745 days). Thirteen patients (31.0%) did not receive RT due to early progression (n=8), treatment-related death (n=3), or physician’s discretion (n=2). As of 2011, RT timing changed from delayed RT (pre-2011) to early adjuvant RT (post-2011). As a result, the median period between diagnosis and RT was significantly shortened from 314.9 days in the pre-2011 group to 159.0 days in the post-2011 group (p=0.04). The period between diagnosis to RT was 114.6±104.8 days in patients who received early adjuvant RT and 313.4±231.2 days in patients who received RT as salvage therapy (p < 0.01).Among 29 patients who received RT, early adjuvant RT was administered in 14 patients (2 in the pre-2011 group and 12 in the post-2011 group) (Table 3). They received local RT at a dose of 36–63 Gy after surgery. Of them, five patients received additional CSI at a dose of 23.4–30.6 Gy concurrently (n=2) or at 3 years of age (n=3) due to M+ disease at diagnosis. Thirteen patients received local RT as salvage therapy at a dose of 25.2–55.8 Gy. Of them, five patients received CSI at a dose of 19.5–23.4 Gy. Two patients received CSI at 3 years of age after completion of HDCT as scheduled.
Table 3
Treatment and clinical outcomes
![]()
Twenty-four patients received HDCT/ASR (15 patients received tandem HDCTs and nine patients received only 1 HDCT). Reasons not having second HDCT included physician’s discretion (n=4), PD or relapse (n=3), prolonged bone marrow suppression (n=1), and patient refusal (n=1). During the first HDCT, eight patients received conditioning regimen including topotecan, thiotepa, and carboplatin instead of CTE. During the second HDCT, two patients received conditioning regimen including busulfan, melphalan, and thiotepa instead of CM.
Significantly more patients who received early adjuvant RT proceeded to HDCT compared to patients who did not (78.6% vs. 46.4%, p=0.04). For patients not receiving HDCT (n=18), reasons included PD or relapse (n=12), treatment-related death (n=4), patient refusal (n=1), and death due to unknown cause (n=1). The median age at the time of the first HDCT was 21.0 months (range, 9.1 to 44.5 months). The median time between diagnosis and the first HDCT was 7.5 months (range, 5.3 to 16.1 months). Disease status before the first HDCT was CR in 16 and PR in eight. Among 15 patients who underwent a second HDCT, disease status before the second HDCT was CR in 10, PR in four, and PD in one. The median interval between the first and second HDCT was 91 days (range, 35 to 126 days).
3. Clinical course and survival
Excluding one patient who received palliative care only, data for 42 patients were analyzed (Table 3). With a median follow-up of 90 months (range, 27 to 172 months), 27 patients (64.3%) showed at least one episode of PD. The first date of PD was at 160 days (median; range, 13 to 585) after the diagnosis. Twenty-one of 27 patients who showed PD died due to PD at a median of 4 months (range, 0.1 to 5.4 months) from the first day of documentation of PD. Immediate salvage therapy consisted of surgery in 11, chemotherapy in four, RT in four, gamma knife surgery in one, and supportive care only in seven. PD was found within 2 months of resection surgery before initiation of chemotherapy in three, during chemotherapy in 14, during adjuvant RT in one, during HDCT in two, and after completion of HDCT in seven. Among 14 patients with PD found during induction chemotherapy, 13 did not receive adjuvant RT previously while one patient had received RT previously (p < 0.01). Of nine patients who experienced relapse during or after HDCT, seven patients died due to PD. Two patients are still alive without disease. The median time between HDCT and subsequent relapse was 3 months (range, 1 to 11 months).
The 1- and 3-year PFS is 51.2% and 28.5%, respectively. The 1- and 3-year OS is 61.9% and 38.1%, respectively (Fig. 2). Of 42 patients analyzed, 27 patients died due to PD (n=21), treatment-related toxicity (n=5), or unknown causes (n=1).
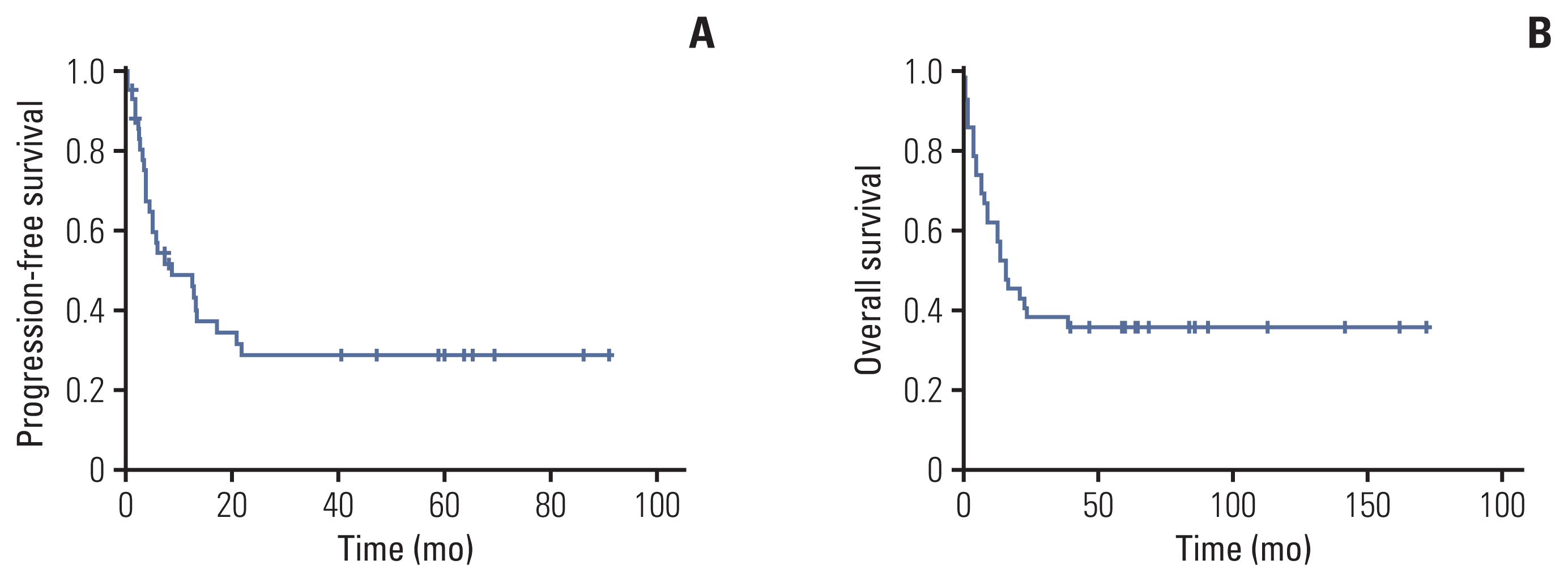
At the last follow-up, 15 patients were alive. All surviving patients received HDCT/ASR. Patients who achieved CR before the first HDCT showed significantly higher 3-year OS compared to patients who did not (81.3% vs. 37.5%, p < 0.01). There was no significant difference in PFS or OS between patients who underwent 1 HDCT and those who underwent 2 HDCTs.
4. Risk factors
Results of univariate and multivariate analyses for PFS are shown in Table 4. In univariate analysis, factors associated with higher 3-year PFS were no metastases (p=0.03), diagnosis after 2011 (p=0.04), early adjuvant RT (p < 0.01) and HDCT/ASR (p < 0.01) (Fig. 3). Age at diagnosis (p=0.53) and extent of resection (p=0.29) failed to influence survival rate in our study. Among 14 patients who received adjuvant RT, only three patients showed PD during (n=1) or after (n=2) the planned RT, while 24 of 28 patients who did not receive adjuvant RT showed PD (p < 0.01). Among 18 patients with metastases, patients who received early adjuvant local RT showed significantly higher PFS and OS compared to those who did not (3-year PFS: 75.0% vs. 0%, p=0.04; 3-year OS: 75.0% vs. 14.3%, p=0.03).
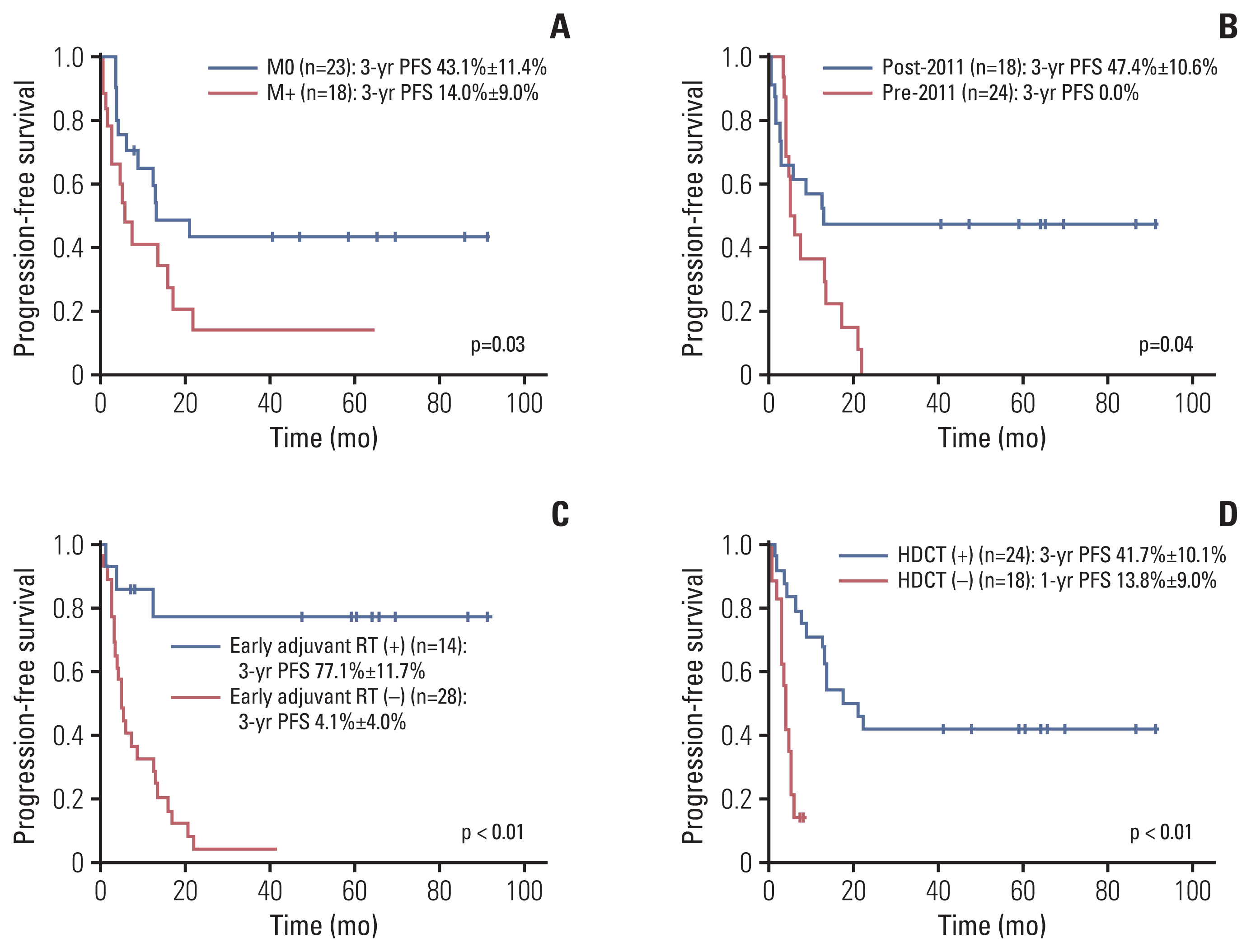 | Fig. 3Progression-free survival according to metastases (A), year of diagnosis (B), early adjuvant radiation (C), and high-dose chemotherapy (D).
|
Table 4
Univariate and multivariate analysis of the probability of progression-free survival
| Univariate | Multivariatea) | |||
|---|---|---|---|---|
|
|
|
|||
| HR (95% CI) | p-value | HR (95% CI) | p-value | |
| Years of diagnosis | ||||
|
|
||||
| Pre-2011 | 1 | 0.04 | 1 | 0.57 |
|
|
||||
| Post-2011 | 0.47 (0.23–0.96) | 0.81 (0.44–1.62) | ||
|
|
||||
| Patient age (yr) | ||||
|
|
||||
| < 1 | 1 | 0.53 | ||
|
|
||||
| ≥ 1 | 0.79 (0.39–1.59) | |||
|
|
||||
| Metastases | ||||
|
|
||||
| Yes | 1 | 0.03 | 1 | 0.53 |
|
|
||||
| No | 0.46 (0.23–0.90) | 0.85 (0.43–1.59) | ||
|
|
||||
| Extent of resection | ||||
|
|
||||
| GTR | 1 | 0.29 | ||
|
|
||||
| No GTR | 1.23 (0.56–2.27) | |||
|
|
||||
| Early adjuvant RT | ||||
|
|
||||
| Yes | 1 | < 0.01 | 1 | < 0.01 |
|
|
||||
| No | 5.68 (2.14–15.12) | 6.42 (2.29–17.90) | ||
|
|
||||
| HDCT | ||||
|
|
||||
| Yes | 1 | < 0.01 | 1 | < 0.01 |
|
|
||||
| No | 10.26 (3.81–27.66) | 12.0 (3.91–37.28) | ||
![]()
In multivariate analyses, the use of HDCT/ASR and early adjuvant RT remained significant prognostic factors for PFS (both p < 0.01). For OS, the use of HDCT/ASR was the only significant prognostic factor (p < 0.01). Among patients who received both early adjuvant RT and HDCT/ASR, 3-year PFS and OS were 81.8% and 90.9%, respectively.
5. Toxicity
During chemotherapy, the most frequently reported significant toxicities were bone marrow suppression and febrile neutropenia followed by infection, gastrointestinal disturbances, electrolyte disturbances, and hepatic disturbances. During HDCT, infection was the most common toxicity. Bacteremia was detected during six of 39 HDCT (3 Staphylococcus epidermidis, 2 Pseudomonas aeruginosa, and 1 Bacillus species), five of which occurred during the second HDCT course. One patient experienced sinusoidal obstruction syndrome during their second HDCT. There were five toxic deaths due to sepsis: four occurred under chemotherapy and one occurred following the second HDCT. To date, second malignancies have not been reported.
Go to :
Discussion
This is the most recent update of Korean ATRT patients under 3 years of age. Tumors progressed or relapsed in 27 of 42 patients at a median of 160 days from diagnosis in our study. The 3-year PFS and OS were 28.5% and 38.1%, respectively. The 3-year PFS was improved from 0.0% in pre-2011 to 47.4% in post-2011. At diagnosis, metastasis was the only characteristic among several disease characteristics known to affect survival.
Our analysis highlights several points of interest for this young age group. First, our data suggest the importance of early adjuvant RT. We found a survival benefit associated with early adjuvant RT. Traditionally, RT has been deferred or omitted because of the risk of adverse effects, especially for patients under 3 years of age [12]. However, more recent data suggest that RT might be more efficacious than chemotherapy for ATRT patients, even for very young children [2,13,14]. In a previously reported Korean study that included patients treated with tandem HDCT/ASR, all surviving patients received RT at an early stage in their treatment [15]. No patients who received induction chemotherapy and HDCT/ASR without RT survived. This suggests that HDCT/ASR cannot replace RT for local control. Similarly, we found that patients who received early adjuvant RT showed a significantly lower chance of having PD during induction chemotherapy and a higher chance to proceed to HDCT compared to patients who did not, thus leading to improved survival. For patients with M+ disease at diagnosis, adjuvant local radiation significantly improved survival in the current study. Overall, local RT should be considered earlier in therapy to improve survival.
Secondly, our data suggest that HDCT is associated with better survival. For patients who received HDCT, PFS and OS were significantly higher compared to those who did not. There has been no randomized study to evaluate the efficacy of HDCT/ASR in ATRT patients under 3 years of age. A few studies have suggested that HDCT might have a favorable effect on ATRT patients [14,16]. A recent Canadian study has reported better 5-year PFS in patients with ATRT under 1 year of age receiving HDCT group compared to those without receiving HDCT (50.1% vs. 11.3%, p < 0.001) [17]. However, due to small numbers of patients included and various treatment modalities used, no firm conclusions can be made regarding the role of HDCT/ASR in ATRT patients. In this study, all surviving patients received HDCT/ASR. However, this does not reflect the definite efficacy of HDCT/ASR. Considering that patients who achieved CR before their first HDCT showed significantly higher OS than those who did not, multimodal treatment that might lead to CR and additional HDCT might result in better outcomes. In this study, patients who achieved CR before the first HDCT showed significantly higher OS. This suggests that pre-HDCT tumor status is important for predicting outcome. Thus, careful consideration is required when selecting candidate patients for HDCT/ASR.
Importantly, we should consider the likelihood of selection bias toward “favorable cases” in patients subjected to HDCT, which could be associated with molecular distinction. Recently, Reddy et al. [18] have reported that patients with group 1/SHH-NOTCH tumors have less aggressive feature. Biological investigation of ATRT should be continued and subsets of patients who can be cured with less therapy may exist [19].
The optimal combination of regimens for tandem HDCT has not been determined. Rosenfeld et al. [12] have reported the feasibility of tandem HDCT/ASR in patients with brain tumors using CTE-CM. Although they concluded that the CTE-CM regimen was not feasible due to toxicity, tandem CTE-CM HDCT/ASR was feasible in our study. Toxicities in our present study were manageable and only one toxic death associated with HDCT occurred. This could be due to the fact that patients were given a sufficient rest period between the first and the second HDCT/ASR. Sung et al. [20] have reported that a shorter interval (< 12 weeks) between the first and second HDCT/ASR is associated with higher TRM. Another thing to be noted was that there was no significant survival difference between patients who received 1 HDCT and those who received 2 HDCTs. In addition, bacteremia occurred more frequently in the second HDCT than in the first HDCT. Randomized trials with larger cohorts are needed to determine whether the possible survival benefit of tandem HDCT/ASR over single HDCT/ASR might ultimately outweigh adverse effects associated with dose intensive tandem HDCT/ASR.
Thirdly, optimal induction chemotherapy should be explored. Efficacies of many different treatments have been explored. However, there is no consensus regarding standard chemotherapy for ATRT. Due to the desire to avoid unacceptable adverse effects of RT on the developing brain, many institutions adopt chemotherapy-based strategies designed to avoid or delay RT. Despite often impressive responses to chemotherapy, the majority of patients in many published studies developed progressive disease early, suggesting a rapid development of resistance of ATRTs [10,21,22]. Similarly, in our study, PD occurred during induction chemotherapy in about half of cases. Considering that the most common reason not having HDCT/ASR was PD during induction chemotherapy, we could consider shorten the current six cycles of induction chemotherapy. Furthermore, patients who received induction chemotherapy without early adjuvant RT showed significantly lower survival compared to patients who received early adjuvant RT. This suggests that CECV and CEIV chemotherapy regimen used in our patients might be insufficient to prevent tumor progression/relapse. Recently, ACNS0333 comprised with three cycles of chemotherapy which incorporated high dose MTX showed promising result, with 4-year OS of 43% [18]. High-dose MTX used in “Head Start II” also appears to be efficacious and well tolerated in ATRT [8]. Slavc et al. [23] have also demonstrated the efficacy of high-dose MTX in ATRT patients. Besides intensification using cytotoxic agent, there are increasing data suggesting that ATRT might be a good candidate for pathway-specific targeted therapies, some of which are currently used in clinical trials, including AURKA, EZH2, and CDK4/6 inhibitors [24,25]. Optimwal combination of cytotoxic agents and targeted inhibitors should be explored to prevent early progression of ATRT.
Lastly, we found that the post-2011 group had significantly better PFS than the pre-2011 group. The major difference between pre- and post-2011 protocols was adjuvant local radiation and IT chemotherapy in the post-2011 protocol. There was no difference in induction chemotherapy or HDCT between pre- and post-2011 protocols. We believe that the post-2011 protocol with a combination of adjuvant RT and IT chemotherapy might have prevented early progression, eventually improving the outcome as reported by Chi et al. [10]. The role of adjuvant RT has already been described above. IT therapy was incorporated as a method of providing prophylaxis and/or treatment to the CNS axis in the post-2011 protocol. Whether IT therapy could substitute for cranial irradiation for CNS treatment and/or prophylaxis was unclear because both modalities were used in the post-2011 protocol. Considering that the year of diagnosis (pre- vs. post-2011) was not a significant predictor of survival in multivariate analyses, IT therapy might not have a beneficial role in improving survival. However, in many studies, IT chemotherapy has shown potential benefit as an addition to local RT or to intensify therapy in patients who are not candidates for CSI [10,26]. IT chemotherapy shows good penetrance into the CSF. Thus, it might be efficacious in eradicating ATRT cells in CSF, allowing postponement of CSI for patients who receive focal irradiation only. A meta-analysis by Athale et al. [26] has shown that IT therapy leads to a significantly higher OS. A more detailed randomized study I needed to evaluate IT therapy in infant ATRT to define role of IT therapy in these patients.
This study has some limitations. First, this study had a non-randomized and retrospective design. Second, we did not collect data for germline mutations in SMARCB1/INI1. Therefore, we could not assess the association between germline status and outcome. No molecular profiling was included, which could be potential prognostic indicators. Third, because current multimodality treatment strategies for ATRT include brain RT, limited data on neurocognitive outcomes of survivors raise a significant concern [27]. Although data stress the importance for RT in younger age group, this treatment option comes at a cost of serious long-term sequelae such as cognitive, motor, visual, and hearing impairment [28,29]. In addition, combining multimodal treatment deserves attention. Comprehensive long-term follow-up neuropsychologic assessments are planned for our surviving cohort. Lastly, as described above, a more detailed analysis is needed in the future to identify the role of IT therapy in the prevention and treatment of metastases.
In summary, aggressive therapy including early administration of local RT and HDCT/ASR, which was adopted in KSPNO-S1102, should be considered to improve outcomes of ATRT in children below the age of 3 years. Despite the high probability of early PD in ATRT, for patients who received both early adjuvant RT and HDCT/ASR, OS exceeded 90%. Further clinical trials may be required to determine optimal adjuvant treatments such as RT field and intensity of HDCT/ASR and the role of IT chemotherapy for patients with ATRT. Future studies regarding molecular characterization of ATRT and its prognostic implication might change current treatment strategies and delineate the group of patients so that treatment intensity could be reduced.
Go to :
 XML Download
XML Download