

 PDF
PDF Citation
Citation Print
Print


Introduction
Fibrolamellar hepatocellular carcinoma (FLHCC) is different from conventional hepatocellular carcinoma (HCC) [1]. The incidence of FLHCC is very low, and it affects adolescents and young adults without any pre existing liver disease [1]. Recent studies suggest that FLHCC is an independent entity, showing distinctive molecular profile [2]. Several case reports described paraneoplastic syndrome of FLHCC with symptoms such as hyperammonemia [3–6], gynecomastia [7], and venous thrombosis [8]. Hyperammonemic encephalopathy (HAE) is the most serious paraneoplastic syndrome. However, owing to its rarity, no management guidelines exist for cancer-related HAE. Herein, we report a case of an 18-year-old male patient with FLHCC who developed HAE during the first course of chemotherapy. He was successfully treated with hemofiltration, sodium benzoate, and sodium phenylbutyrate. He underwent surgery and successive chemotherapy, and his ammonia levels were maintained in the normal range without ammonia scavenger therapy.
Case Report
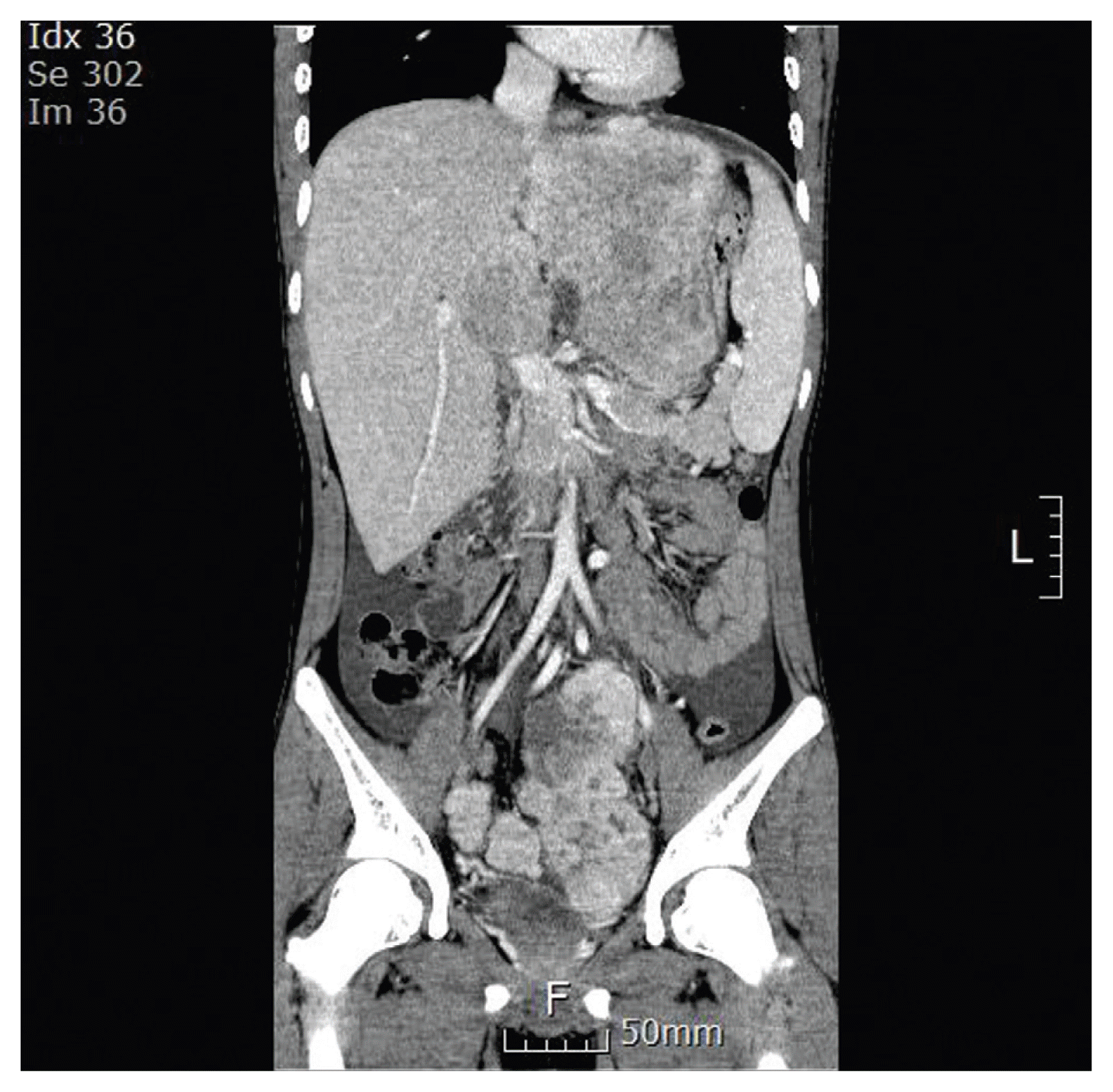
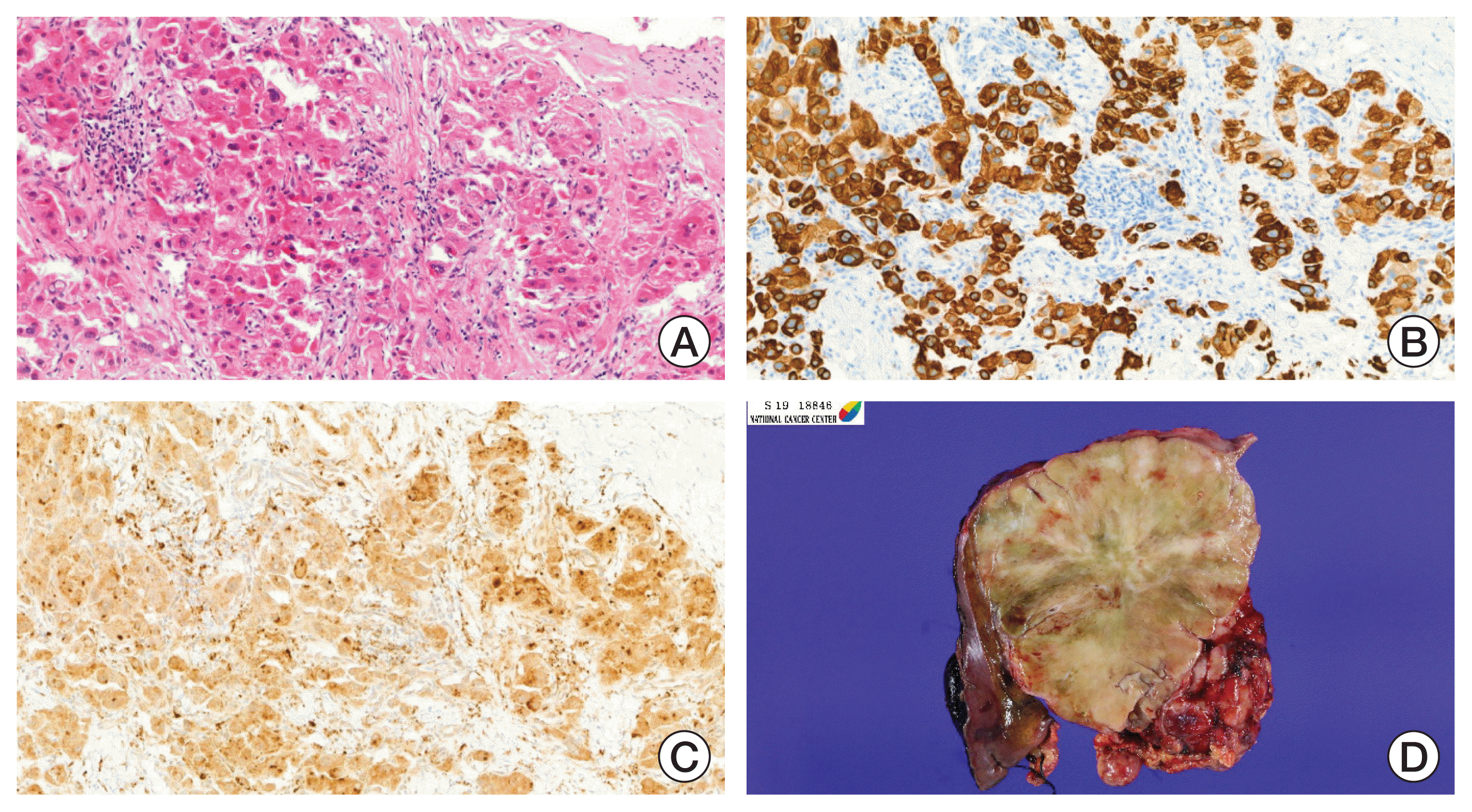
An 18-year-old boy presented with a history of fatigue, vomiting, abdominal pain, and distension. Computed tomography (CT) revealed a huge mass (13.7×10.7 cm) occupying the left liver and two masses (8.7×7.3 cm, 3.9×2.9 cm) in the pelvic cavity. Multiple lymph nodes were enlarged in right anterior and posterior cardiophrenic angles (Fig. 1). An initial laboratory investigation showed normal bilirubin, slightly elevated aminotransferase levels (aspartate aminotransferase [AST], 85 U/L; alanine aminotransferase [ALT], 154 U/L) and α-fetoprotein (27.2 ng/mL). He was previously healthy with no signs of chronic liver disease and had negative results on serologic testing for hepatitis B (hepatitis B virus surface antigen, hepatitis B virus surface antibody) and hepatitis C (anti–hepatitis C virus). He underwent fine needle biopsy and was diagnosed with FLHCC (Fig. 2). Considering the extent of the tumor, the patient was not considered a candidate for surgical resection. Systemic chemotherapy was initiated with cisplatin (80 mg/m2 on day 1), doxorubicin (30 mg/m2 on days 1 and 2), 5-fluorouracil (5-FU; 400 mg/m2 on days 1–4), and interferon-α (5×108 U/m2 on days 1–4). At midnight on day 3 of chemotherapy, the patient became agitated and was walking and running around the ward, not responding to verbal stimuli. Laboratory testing revealed a serum ammonia level of 393 μg/dL, AST/ALT level of 201/126 U/L, and total bilirubin level of 0.3 mg/dL. Head CT did not reveal any significant findings. He was diagnosed as HAE, and chemotherapy (4th dose of 5-FU and interferon-α) was stopped. Treatment with rifaximin and lactulose was initiated. However, his serum ammonia level rapidly increased to 500 μg/dL, and his mental status deteriorated to the semicoma state.
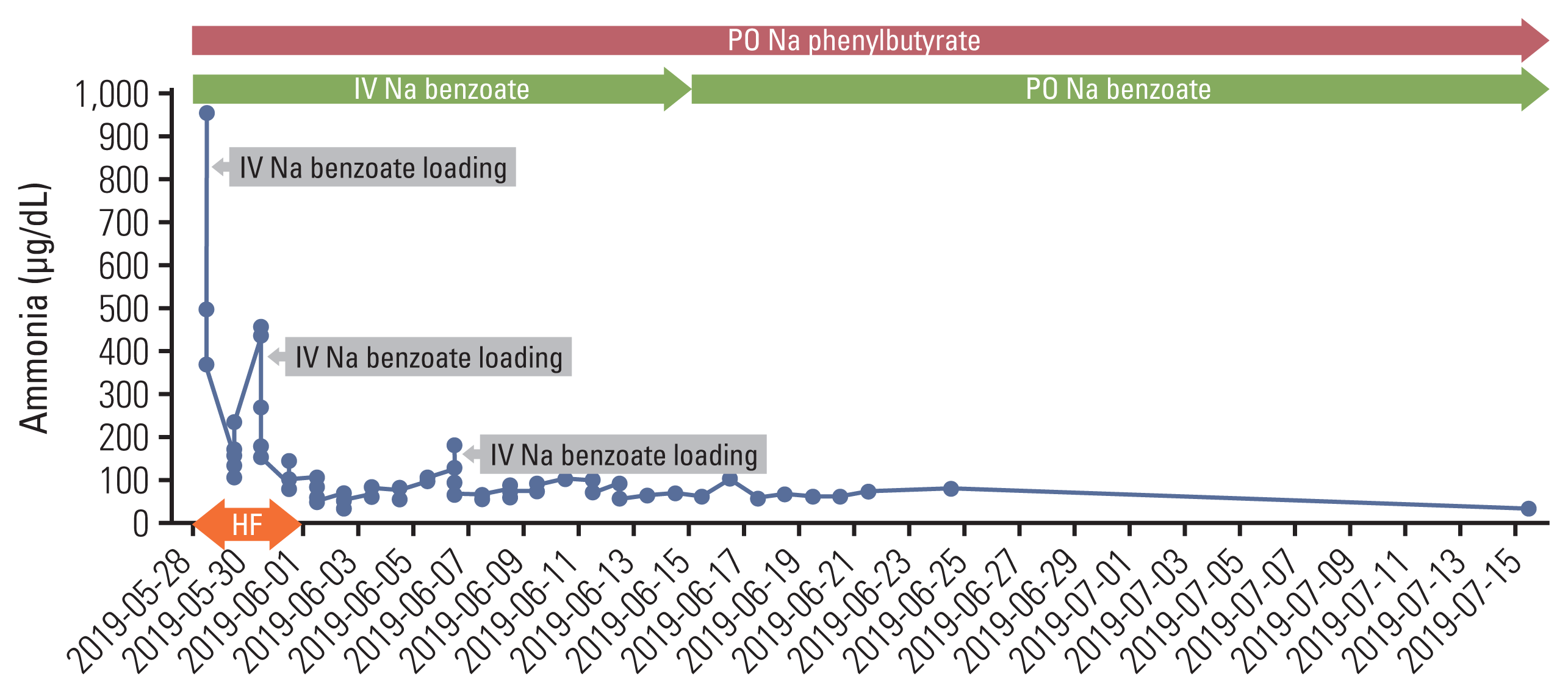
The patient was transferred to the intensive care unit (ICU) and was on mechanical ventilator support under sedation using midazolam, remifentanil, and vecuronium. The metabolic and genetics department was consulted, and treatment for hyperammonemia was started as follows: (1) continuous venovenous hemofiltration, (2) intravenous infusion of sodium benzoate at a loading dose of 8.8 g (170 mg/kg), followed by continuous infusion (8.8 g/day) and sodium phenylbutyrate (5 g four times a day, via Levin tube), (3) 20% dextrose for caloric intake (6 mg/kg/min), and (4) administration of lactulose and rifaximin via Levin tube. His ammonia level decreased to 164 μg/dL but rebounded to 442 μg/dL on the third day of ICU care. Sodium benzoate (8.8 g) was loaded again, and his ammonia level decreased to 135 μg/dL. On the sixth day of the above-mentioned treatment, continuous venovenous hemofiltration was discontinued. On the eighth day, the patient was extubated, and sodium benzoate/sodium phenylbutyrate administration was changed to an intermittent schedule, with sodium benzoate 1.8 g administered as intravenously every 8 hours, sodium phenylbutyrate 3 g four times a day, and arginine 2.5 g twice a day. He was started on a low-protein diet (1 g/kg/day) through a Levin tube. Two days later (10th day of ICU care), he became disoriented and could not properly respond to verbal stimuli, with an ammonia level of 173 μg/dL. The low-protein diet was discontinued, and a continuous infusion of sodium benzoate (5.5 g/day, after a loading dose of 5.5 g) was started again, as well as citrulline (5 g three times a day). There were no focal neurologic signs, and brain magnetic resonance imaging (MRI) did not reveal any abnormality. As the serum ammonia level started decreasing (59 μg/dL), his conscious level improved. On the 12th day, he started Levin tube feeding and oral medications (sodium phenylbutyrate 6 g/day, L-carnitine 2,550 mg/day); citrulline was discontinued. On the 14th day of ICU care, sodium benzoate infusion was changed to an intermittent schedule (1.8 g every 8 hours). He resumed the low-protein diet (0.5 g/kg/day), and his ammonia level was maintained in the range of 90–100 μg/dL. He was transferred to a general ward on the 16th day of ICU care, and sodium benzoate was changed to an oral form (3 g three times a day). With sodium benzoate, sodium phenylbutyrate (3 g three times a day), arginine (2.5 g twice a day), and L-carnitine (660 mg once daily), his ammonia level further decreased to 50–60 μg/dL (Fig. 3). On the ninth day at the general ward, laboratory analyses were performed to identify the underlying mechanism of his HAE. The patient’s serum amino acid analysis revealed the following findings: decreased citrulline and ornithine, normal alanine and glutamine. His urine orotic acid level was elevated, and serum acylcarnitine was slightly elevated. These results suggested impaired activity of ornithine transcarbamylase (OTC). However, polymerase chain reaction (PCR) and direct sequencing of the OTC gene did not reveal any pathogenic variant, deletion, or duplication.
On the 20th day at the general ward, the patient underwent a second course of chemotherapy involving cisplatin+doxorubicin+5-fluorouracil+interferon-α. One month later, he underwent extended left hemihepatectomy en bloc with the caudate lobe, excision of pelvic mass, and peritoneal seeding. Extensive lymph node dissection was also performed at the hepatoduodenal, cardiophrenic, para-aortic, and pelvic lymph nodes. Immunohistochemical staining confirmed a profile consistent with that of FLHCC, that is, positive for cytokeratin 7, glypican 3, and CD68. We did not test for the DNAJB1-PRKACA fusion. Next-generation sequencing (NGS) was performed to identify actionable therapeutic targets but only revealed APC mutation (c.4666delA, p.T1556fs), with an allele frequency of 15.53%. His postoperative period was uneventful and the ammonia level further decreased (30–40 μg/dL). One month after the surgery, treatment for hyperammonemia was discontinued. He received two additional courses of chemotherapy involving cisplatin+doxorubicin+5-FU+interferon-α. However, CT and MRI obtained 3 months after the surgery revealed enlarged left external iliac lymph nodes and multiple enhancing nodules at the perihepatic, perigastric, mesentery, omentum, and pelvic cavity. The chemotherapy regimen was changed to vincristine (1.5 mg/m2 on days 1, 8)+irinotecan (50 mg/m2 on days 1–5) and then to gemcitabine (1,000 mg/m2 on day 1)+cisplatin (20 mg/m2 on days 1–5). CT performed after three courses of gemcitabine+cisplatin showed a decrease in the size of the peritoneal nodules and iliac lymph node. Furthermore, the patient experienced severe, recurrent abdominal pain with vomiting, and CT images suggested obstructive ileus. Exploratory laparotomy was performed, and multiple adhesive bands were dissected off the small intestine and excised. Additionally, two metastatic nodules were resected. At the time of this report writing, the patient had just completed the sixth course of gemcitabine-cisplatin, and his disease was stable. He is on normal regular diet and his ammonia level remains normal (30–40 μg/dL).
Discussion
We report an 18-year-old male patient with advanced FLHCC who developed HAE during the first course of chemotherapy comprising o cisplatin, doxorubicin, 5-FU, and interferon-α. His HAE was successfully treated with continuous venovenous hemofiltration, ammonium scavengers, and amino acid supplementation. Serum amino acid and urine organic acid analyses suggested urea cycle dysfunction. However, PCR and direct sequencing of the OTC gene did not reveal any pathogenic variants. Intriguingly, APC gene mutation was detected from NGS of the tumor tissue, suggesting a possible relation to the development of HAE. After tumor excision, serum ammonia levels were maintained in the normal range without treatment with ammonium scavengers.
HAE in FLHCC was first reported in 2009 [3]. This condition is very rare, and approximately 10 cases are reported thus far; many of the patients present HAE at the time of diagnosis of FLHCC [3–6]. One of them recovered from HAE after liver transplantation [4]. Similar to our case, a 31-year-old man developed HAE on the third day of gemcitabine+oxaliplatin chemotherapy [5]. Several mechanisms were proposed on the development of HAE in patients with FLHCC. Some studies suggest that urea cycle dysfunction is caused by an imbalance in the activities and ratios of OTC and ornithine decarboxylase (ODC) [6]. Reduced activity of OTC or augmented activation of ODC results in ornithine consumption and urea cycle disturbance, thereby causing hyperammonemia [6,9]. A recent study suggested that the molecular pathogenesis of FLHCC may contribute to urea cycle dysfunction [6]. Heterozygous deletion of chromosome 19 results in chimeric DNAJB1-PRKACA kinase, which is frequently identified in FLHCC [10]. This chimeric DNAJB1-PRKACA kinase increases the expression of Aurora kinase A, leading to the overexpression of c-Myc that upregulates ODC function [6,10].
The precise mechanism of the development of HAE in our case is unclear. Initially, there were no specific findings in his metal or neurologic status, and his serum ammonia level was not tested. He developed HAE on the third day of chemotherapy, which suggests a causal relationship between chemotherapeutic agents and HAE. There are case reports about chemotherapy-associated hyperammonemia. For example, patients with acute lymphoblastic leukemia receiving corticosteroids [11] and L-asparaginase [12] show a transient increase in the serum ammonia level. Several case reports described HAE in patients receiving 5-FU, cytarabine, cyclophosphamide, oxaliplatin, vincristine, etoposide, anthracycline, busulfan, methotrexate, topotecan, gemcitabine, and tyrosine kinase inhibitors [5]. Our patient received steroids (as antiemetics) and 5-FU. Another culprit that might have contributed to HAE is APC tumor suppressor gene mutation identified by the NGS assay of the tumor. Unfortunately, our case was not tested for heterozygous deletion of chromosome 19 or chimeric DNAJB1-PRKACA kinase. APC mutation was identified by NGS assay of the tumor with an allele frequency of 15.53%. Several in vitro studies suggest that APC mediates ODC expression through a c-Myc– dependent mechanism, thereby affecting ODC transcription [13]. Tumor cells with truncated APC exhibit high levels of β-catenin, leading to increased expression of c-Myc [13]. We presume that both APC mutation and chemotherapy might have caused HAE resembling OTC deficiency.
There are no management guidelines for cancer- or chemotherapy-associated hyperammonemia or HAE. Therefore, treatment of hyperammonemia or HAE is extrapolated from that for hepatic encephalopathy or OTC deficiency [14]. Clinical features of hyperammonemia can deteriorate from irritability, confusion to seizure, coma, and even death [14]. Ammonia scavenger therapy should be initiated when ammonia levels are greater than 100 μg/dL [5]. When the serum ammonia level is extremely high (500 μg/dL), dialysis/rapid hemofiltration should be started immediately [5]. In our case, the patient developed HAE during the first course of chemotherapy. Ammonia scavenger therapy normalized his ammonia level and was maintained until the second course of chemotherapy and surgery. After surgery, his ammonia level remained normal without ammonia scavenger therapy, suggesting that his HAE was a paraneoplastic phenomenon of FLHCC.
While HCC is prevalent in Asia, the incidence of FLHCC is similar across different ethnic groups [1]. FLHCC comprises less than 1% of all primary liver cancer cases in the United States [1]. Recent studies suggest that FLHCC has a distinctive molecular profile, with focal deletion leading to DNAJB1-PRKACA gene fusion [2]. There are limited studies on the treatment outcome of patients with FLHCC. Despite the large tumor size and advanced stage at presentation, FLHCC patients exhibit significantly better 5-year relative survival than those with conventional HCC [1]. It is unclear whether biologic difference or absence of pre existing liver disease in FLHCC attributes to better survival. Except for surgery, no consensus exists for the treatment of FLHCC. Chemotherapy regimens for unresectable cases include cytotoxic agents, bevacizumab, and tyrosine kinase inhibitors [5]. A phase II trial involving nine cases of FLHCC suggested a potential benefit of 5-FU and interferon [15]. Our patient received perioperative chemotherapy including 5-FU and interferon. Generally, surgical resection is not considered for case like ours, even after systemic chemotherapy. However, there are several reports favoring aggressive cytoreductive surgery prolongs the survival of HCC patients with peritoneal metastases [16,17]. Our case received surgery for the following reasons: (1) the patient was young without any underlying disease, (2) tumors, including liver mass, peritoneal and pelvic metastases appeared to be feasible for resection without compromising patient’s safety, (3) to alleviate clinical symptom (HAE, early satiety, nausea, abdominal discomfort and pain) caused by the multiple large tumors. In the absence of any evidence-based treatment options for aggressive FLHCC, disappearance of HAE after surgery showed that our decision was appropriate. However, the size of the peritoneal nodule increased 3 months after the surgery and combination chemotherapy. The chemotherapy regimen was changed, and the patient was on third-line chemotherapy consisting of cisplatin and gemcitabine, and the patient has been alive for 12 months since the diagnosis of FLHCC.
In conclusion, a young boy with FLHCC presenting HAE, which mimicks OTC deficiency, was successfully treated with hemofiltration and ammonia scavenger therapy. Lack of significant findings in OTC gene analysis and the normal ammonia level after surgery imply that FLHCC directly caused urea cycle dysfunction. We assume that APC mutation might be partially related to the development of HAE in our case. Oncologists should be aware about the HAE, a rare but serious metabolic complication of FLHCC. Treatments extrapolated from OTC deficiency, such as continuous venovenous hemofiltration and ammonia scavengers, will be helpful and could partially applied to HAE related to any malignancies.
 XML Download
XML Download