

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Sarcoidosis is a multisystemic disease characterized by noncaseating granulomas, that predominantly affect organs such as the lungs and lymph nodes. The neurological manifestation of sarcoidosis has been described in 5% to 10% of patients [1,2]. Neurosarcoidosis can involve the cranial nerves, meninges, brain parenchyma, spinal cord, dura, muscle, and peripheral nerves. Spinal cord involvement, including intramedullary and extramedullary lesions, is uncommon [3]. Here, we report a patient with cervical myelitis and a previous diagnosis of pulmonary sarcoidosis. His clinical course, magnetic resonance imaging (MRI) findings, and clinical management are described.
CASE REPORT
A 32-year-old male presented with a 1-year history of pain in his neck, arms, and hands. He also complained bilateral numbness of his upper extremities 2 months prior to admission.
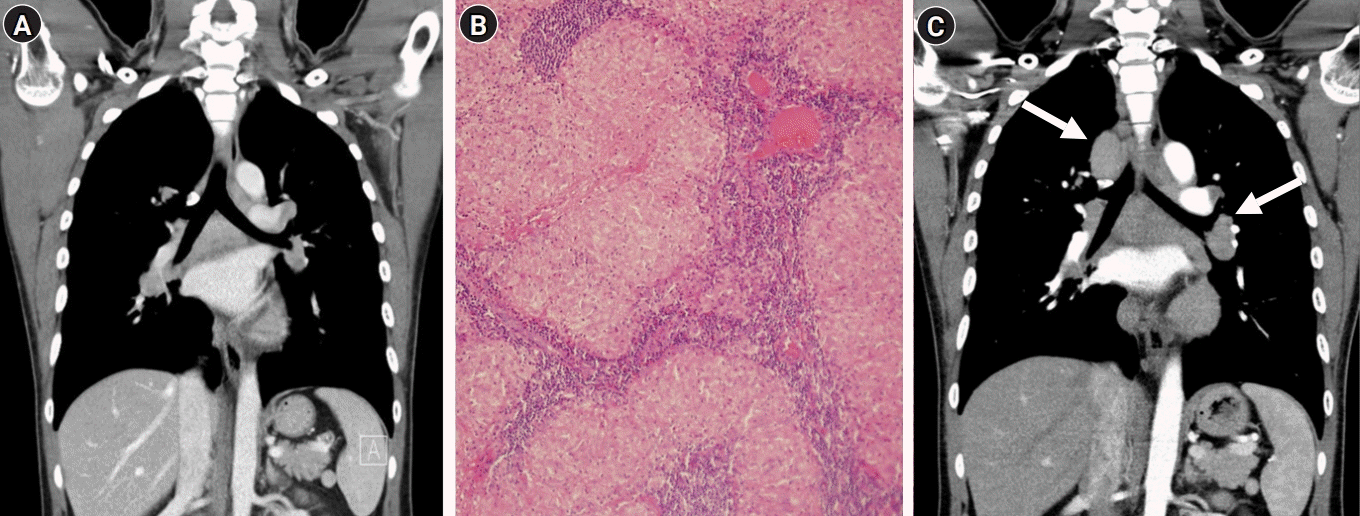
Four years ago, he had no pulmonary symptoms, but bilateral hilar and lower paratracheal lymphadenopathy was incidentally discovered during a routine medical chest radiography and computed tomography (CT) (Fig. 1A). A biopsy via video-assisted thoracoscopic surgery showed noncaseating epithelioid granulomatous lesions compatible with pulmonary sarcoidosis (Fig. 1B). Tissue cultures for mycobacteria and fungus were negative. He was treated with low-dose oral steroids for 2 years but discontinued the medication arbitrarily.
On physical examination, the muscle strength in his limbs was normal. Sensory examination revealed that his light touch was decreased on the medial aspect of the bilateral upper arm, forearm, and hand. Deep tendon reflexes were normal. His visual analog scale (VAS) score was 7.
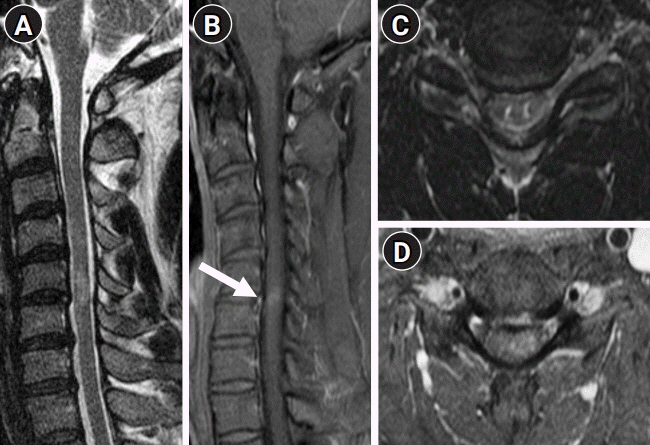
The findings of his nerve conduction studies, electromyography, median, and tibial nerve evoked potential studies we were normal. Brain MRI scan was normal. A spinal cord MRI showed T2 hyperintensity from C5 to C6, with focal patchy gadolinium enhancement between C5 and C6 (Fig. 2). Cerebrospinal fluid (CSF) was acellular with an elevated protein level (52.2 mg/dL). There were no oligoclonal bands in the CSF or serum. The immunoglobulin G (IgG) index was normal (0.52). CSF cytology was negative for malignancy. There were no oligoclonal bands in the CSF or serum. Test results for antinuclear antibody, antineutrophil cytoplasmic antibodies, aquaporin (AQP4) IgG, and myelin oligodendrocyte glycoprotein (MOG) IgG were negative. Thyroid function and the serum IgG4 levels were within the normal range. His serum angiotensin-converting enzyme (ACE) level was elevated (79 U/L [normal range, 12 to 68]). A chest CT revealed increased enlargement of the hilar lymph nodes compared to the previous study (Fig. 1C).
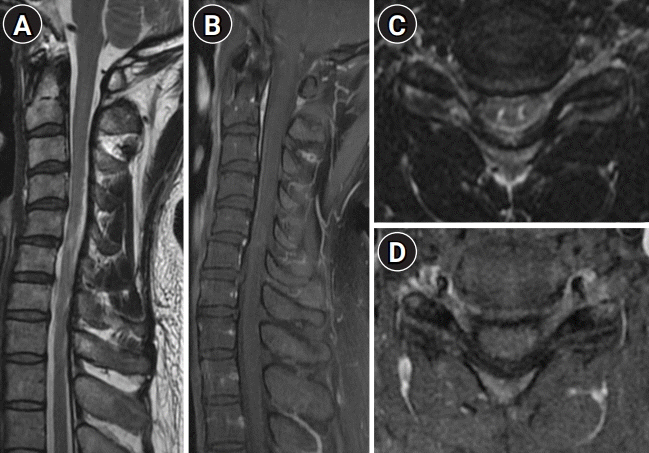
He was diagnosed with a spinal cord manifestation of sarcoidosis and treated with intravenous methylprednisolone (1,000 mg/day, 3 days) followed by oral prednisolone. Azathioprine was administered in addition to prednisolone. At the 2-month follow-up, diminishing enhancement could be seen on the MRI (Fig. 3). The numbness had improved, but mild pain (VAS score, 3) persisted even though he was on continuous oral prednisolone (20 mg) and azathioprine (75 mg) treatment.
This study was approved by the Institutional Review Board of Kosin University Gospel Hospital (KUGH 2020-03-026). Since this was a retrospective case study, the requirement for informed consent was waived.
DISCUSSION
Neurological manifestation occurs in 5% to 10% of patients with sarcoidosis [1,2]. Spinal cord sarcoidosis rarely occurs, and intraspinal sarcoidosis is rarely reported (0.1% to 50% of neurosarcoidosis) [3]. However, neurologic involvement can manifest 2 to 3 years after the development of systemic sarcoidosis [4,5]. Additionally, autopsy studies show that up to half of pathologic neurosarcoidosis cases are not diagnosed during an individual’s lifetime [6]. Therefore, the emergence of neurological symptoms in patients with systemic sarcoidosis should raise suspicion of neural involvement associated with sarcoidosis.
Diagnosis of spinal cord sarcoidosis can be challenging, given that a biopsy of the spinal cord is necessary to establish a “definitive” diagnosis of neurosarcoidosis (Table 1) [7]. Spinal cord biopsies can lead to increased morbidity and mortality [8]. Furthermore, biopsy samples do not always show abnormalities [8-10]. Our case had myelopathy with an elevated CSF protein level, elevated serum ACE level, positive histology of the hilar lymph node, and exclusion of alternative diagnoses, which led to the diagnosis of “probable” neurosarcoidosis [7]. Concerning differential diagnoses, the absence of brain lesions, oligoclonal bands, AQP4, and MOG antibodies and the enlargement of the hilar lymph node on the follow-up chest CT reduced the possibility of a demyelinating disorder of the central nervous system, such as multiple sclerosis and neuromyelitis optica.
In spinal cord sarcoidosis, clinical symptoms and MRI findings depend on the anatomic distribution and stage of the illness. There have been several MRI studies of spinal cord sarcoidosis involving lesions in the thoracic and cervical cord [3,11]. Most MRI findings can be relatively nonspecific, mimicking demyelinating disorders, as well as infectious and neoplastic diseases. An MRI analysis of 16 patients with intramedullary spinal sarcoidosis showed leptomeningeal enhancement, fusiform spinal cord enlargement, focal or diffuse intramedullary disease, and spinal cord atrophy depending on the clinical course [11]. Additionally, gadolinium enhancement occurred in the spinal cord even during the chronic phase of the disease course [11], similar to the heterogeneous intramedullary gadolinium enhancement seen in this case 1 year after symptom onset. This intramedullary T2 high signal intensity and faint enhancement without cord swelling may be related to a result of ischemia and disruption of the neural pathways by sarcoidosis in the chronic phase [11].
Our case showed elevated CSF protein without pleocytosis. Lymphocytic pleocytosis (40% to 70%), elevated protein (40% to 73%), positive oligoclonal bands (22% to 55%), and a low CSF glucose (10% to 20%) were reported in the CSF of neurosarcoidosis patients [7,12,13]. Wengert et al. [12] reported that CSF abnormalities in neurosarcoidosis are related with diffuse leptomeningeal enhancement on MRI and disease activity. Therefore, the CSF study may play a crucial role in assessing disease activity as well as a differential diagnosis.
In sarcoidosis, treatment is aimed at remission and symptom relief. Symptoms caused by inflammation are treated with corticosteroids, preferably in combination with immunosuppressants to prevent relapsing chronic sarcoidosis [14]. In this case, 2 years ago, the patient was lost to follow-up while on steroids. At the time of diagnosis of cervical myelitis, numbness recovered after the steroid pulse, but the patient continued to experience pain. At this point, systemic and periodic monitoring is essential to detect any subsequent inflammation to ensure timely treatment.
In conclusion, a high degree of suspicion of neurosarcoidosis is needed to enable early detection and treatment among patients with systemic sarcoidosis. Early detection and treatment will minimize neurological complications.
 XML Download
XML Download