

 PDF
PDF Citation
Citation Print
Print


Introduction
Glioma is the most common malignant tumor in brain and extremely fatal. The survival time of patients is short, and the prognosis is poor. According to the World Health Organization glioma grading classification criteria, gliomas are classified into low-grade glioma (I–II) and high-grade glioma (III–IV) [1]. At present, first-line therapeutic schedule in clinic is surgery resection combined with chemotherapy and radiotherapy [2]. Temozolomide (TMZ) is a conventional chemotherapy drug for glioma clinical therapy since it readily crosses the blood-brain barrier. However, despite its clinical effect, TMZ shows limited efficacy in prolonging the survivals of patients with glioma [3]. Drug resistance is one major factor that leads to the failure of chemotherapy. It is a critical issue for the cancer therapy to eradicate drug resistance and the underlying mechanisms still need to be investigated. How to enhance the drug sensitivity of glioma to TMZ is a very important direction in the treatment of tumor.
Isocitrate dehydrogenase (IDH) including IDH1 and IDH2 are key enzymes in the citric acid cycle. The role of the enzymes is to catalyze the tranformation of isocitrate to α-ketoglutaric acid (α-KG). Research indicates that mutant IDH1 directly converts α-KG to (R)-2HG [4]. 2HG is an oncometabolite that affects chromatin methylation, cellular differentiation and homologous recombination [5]. IDH1 mutations occurred frequently in low-grade gliomas and in most secondary glioblastomas. Clinically, IDH1 mutations were associated with longer overall survival [6]. Research demonstrated that most low-grade gliomas without IDH mutation presented a malignant phenotype that molecularly like glioblastoma [7]. Further analysis indicated that patients with IDH1 mutation and O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation were more sensitive to TMZ treatment in secondary glioblastoma versus IDH1 wild type and MGMT promoter demethylation [8]. Although the relationship between IDH1 mutation and chemosensitivity is increasingly recognized, the molecular mechanism determining the vulnerability of IDH1 mutation remains to debate.
As clinical first-line drugs for glioma chemotherapy, TMZ is a DNA-methylating agent that indirectly induces DNA double-strand breaks (DSB) [9]. Ataxia telangiectasia mutated (ATM) serves as a key controller of cellular responses to DSB by activating multiple signal transduction pathways. Activated ATM phosphorylates several target proteins including the protein kinases checkpoint kinase 2 (CHK) 1, CHK2, and p53. These proteins are involved in cell cycle, DNA repair, and apoptosis [10]. DNA repair signal pathway can make tumor cells survive from DNA damage caused by chemotherapy. It is an integral part of cancer treatment on account of DNA damage in chromosomal DNA, including radiotherapy and cytotoxic chemotherapy [11]. The combination of inhibitors for specific DNA repair pathways and chemotherapy drugs for DNA damage deserves further investigation.
In the research, we established glioma cell lines that expressed the IDH1-R132H and wild-type control. We found that TMZ inhibited glioma cells proliferation more significantly in IDH1 mutant cells compared to wild type. A lower dose of TMZ was effective to induce cytotoxic damage in IDH1-R132H mutant cells. The expression of γH2AX in IDH1-R132H mutant group was higher than wild-type group suggesting that TMZ induced more DNA damage in IDH1 mutant gliomas. To detect the annexin V/propidium iodide (PI) staining by flow cytometry we demonstrated that IDH1 mutation enhanced TMZ induced apoptosis. The TMZ induced activation of ATM and its substrates CHK2 and p53 were inhibited in IDH1-R132H mutant group. Furthermore, ATM inhibitor enhanced the cytotoxic effects of chemotherapy in IDH1-R132H mutant glioma cells. Taken together, our findings demonstrate that IDH1 mutations enhance TMZ sensitivity via regulating ATM/CHK2 pathway in glioma and suggest a combination therapeutic method in clinic.
Go to : 
Materials and Methods
1. Tumor cell lines
Human glioma cell lines U87 and Ln229 were purchased from the Chinese Academy of Sciences Cell Bank. We cultured the glioma cells in Dulbecco’s modified Eagle’s medium (Corning, Corning, NY) with 10% fetal bovine serum. We added penicillin (100 U/mL) and streptomycin (100 μg/mL) into the culture medium. Cells were maintained at 37°C in a humidified incubator with 5% CO2.
2. Lentivirus production and stable cell line establishment
We created the glioma cell lines (Ln229 and U87) expressing IDH1-R132H mutation or wild-type control. IDH1-R132H mutation lentivirus and wild-type lentivirus were purchased from Shanghai GeneChem (Shanghai, China). The culture medium containing virus was added to cells. After infection for 48 hours, we discard the supernatant and change fresh culture medium. Treat the cells with puromycin to screen the cells. IDH1-R132H expression was confirmed by observation of green fluorescent protein fluorescent and western blot analysis.
3. Cell proliferation assay
Cell viability was detected by the Cell Counting Kit-8 (CCK-8) assay. Briefly, cells were harvested and counted. We seeded the specific number of tumor cells in 96-well plates. Treat the cells with different concentrations of TMZ or dimethyl sulfoxide control. After a certain time, we added the CCK-8 solution into the wells. Glioma cells were detected at 450 nm of absorbance in a microplate reader.
4. Western blotting
Glioma cells were treated with lysis buffer and centrifuged at 12,000 ×g. We separated the protein by gel electrophoresis. Then we transferred the protein from the gel to the PVDF membrane. Blocking the membrane in 5% nonfat milk at room temperature for one hour. The membranes were incubated with specific antibodies. All antibodies were diluted at specific concentrations given by manufacturer including anti-IDH1-R132H (Dianova DIA-H09, Hamburg, Germany), anti-ATM (Proteintech, Rosemont, IL), anti–p-ATM, anti-CHK2, anti–p-CHK2, anti-p53, anti–p-p53, anti-H2AX, anti-Bax, anti–Bcl-2 (Cell Signaling Technology, Beverly, MA) and anti-GAPDH (Proteintech). Incubating the membranes with secondary antibody. The membranes were treated with ECL and scanned by Gel Doc 2000 (Bio-Rad, Hercules, CA).
5. Immunofluorescence assay
Immunofluorescence assay was performed as previously described [12]. Briefly, glioma cells were fixed in 4% paraformaldehyde and infiltrated by 0.1% Triton. The cells were blocked in 1% bovine serum albumin. Anti-γH2AX antibody (1:200, Cell Signaling Technology) and secondary antibody (1:1,000, Molecular Probes, Eugene, OR) were diluted and incubated with tumor cells. The slides were observed by fluorescence microscope (Nikon, Tokyo, Japan).
6. Flow cytometry apoptosis assay
We used the flow cytometry to check cell apoptosis according to the protocol. The glioma cells were collected and washed by phosphate buffered saline. Resuspend cells in binding buffer and transfer 100 μL to 5 mL culture tube. Add phycoerythrin annexin V and 7-amino-actinomycin D. Incubate the cells for 15 minutes at room temperature in the dark. Add 400 μL binding buffer and analyze by flow cytometry.
7. Immunohistochemistry staining
Tissue sections were fixed with formalin and embedded with paraffin. The slices were labeled with antibodies. Immunostaining intensity and reactivity were examined by Case Viewer after scanning using digital microscope. The expression of proteins was quantified by a 4-value score criterion that consisted with intensity score (0, 1, 2, 3 for none, weak, moderate and strong) and percentage score (0, < 10%; 1, 10–40%; 2, 40–70%; 3, > 70%). The final score was the intensity values plus percentage score.
8. Animal models and in vivo tumor formation
Four to 6-week-old female nude BALB/c mice were used for subcutaneous xenograft models. Glioma cells were subcutaneously transplanted into back flanks of BALB/c nude mice. After transplanted for 4 weeks 20 mg/kg TMZ was administered intraperitoneally into mice for consecutive 7 days. All nude mice were killed in the sixth week.
9. Statistical analysis
We conducted student t test to compare two groups. The difference between groups were calculated by one-way ANOVA analysis by SPSS software ver. 19.0 (SPSS Inc., Chicago, IL). Statistical results with mean±SEM in the experiments were calculated and conducted by GraphPad Prism software (ver. 7.0, GraphPad Software Inc., San Diego, CA). Results were performed at least three independent experiments and at least three technical replicates. p < 0.05 was statistical significance.
Go to :
Results
1. IDH1 mutation inhibited TMZ treated glioma cells proliferation
IDH1 mutant glioma cell lines were established that stably expressed IDH1-R132H in Ln229 and U87 cell lines. IDH1-R132H expression was confirmed by western blot (Fig. 1A). We treated glioma cells with or without TMZ and detected cell proliferation in different time points by CCK-8. The result demonstrated that IDH1 mutant cells were more sensitive to TMZ versus wild type (Fig. 1B and C). Moreover, the proliferation rate of IDH1 mutant cells was slower than that of wild-type cells especially in 72- and 96-hours’ time points. The dose-response curves shifted significantly to the left in IDH1-R132H mutant group (for Ln229 cells, 50% inhibiting concentration [IC50] IDH1WT=399.4 μM, IC50 IDH1R132H=149.2 μM; for U87 cells, IC50 IDH1WT=543 μM, IC50 IDH1R132H=259.7 μM) when treated with TMZ in different concentrations (Fig. 1D and E). The IC50 of TMZ in IDH1-R132H mutant group was significantly reduced than wild-type group suggesting that a lower dose of TMZ is effective inducing cytotoxic damage in IDH1-R132H mutant gliomas (Fig. 1F).
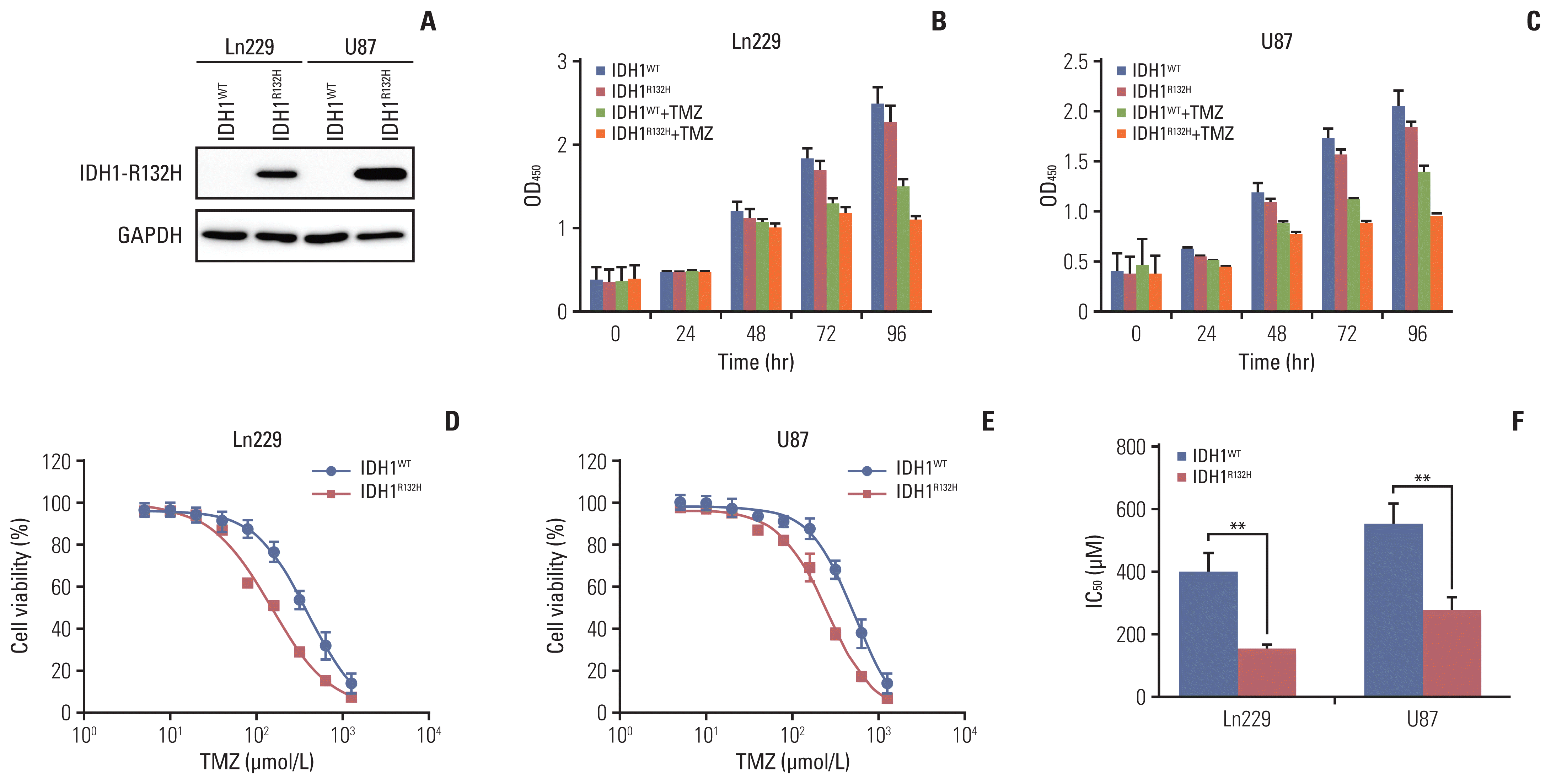 | Fig. 1Isocitrate dehydrogenase 1 (IDH1) mutation inhibits temozolomide (TMZ) treated glioma cells proliferation. (A) IDH1 mutant cells that stably expressing IDH1-R132H mutants in Ln229 and U87 cell lines. IDH1-R132H expression was confirmed by western blot. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (B, C) IDH1 mutant or IDH1 wild-type cells were treated with or without TMZ for 0 to 96 hours. Cell proliferation was detected by Cell Counting Kit-8. (D, E) TMZ dose-response curves of Ln229 and U87 cells with IDH1 mutation for 96 hours. (F) Statistical analysis of IC50 in Ln229 and U87 glioma cells. **p < 0.01.
|
2. TMZ induced more DNA damage in IDH1 mutant glioma
We performed immunofluorescence to examine the DNA damage in glioma cells induced by TMZ. The result demonstrated that the expression of γH2AX in IDH1-R132H mutant cells was stronger than that in the wild-type cells when treated with TMZ (Fig. 2A and B). Quantification of γH2AX in wild-type cells and IDH1 mutant cells treated with TMZ (Fig. 2C and D). In addition, the western blot analysis showed that the expression of γH2AX in IDH1-R132H mutant group was more prominent than that in the wild-type group (Fig. 2E and F). Thus, TMZ induced more DNA damage in IDH1 mutant cells compared with wild-type glioma cells.
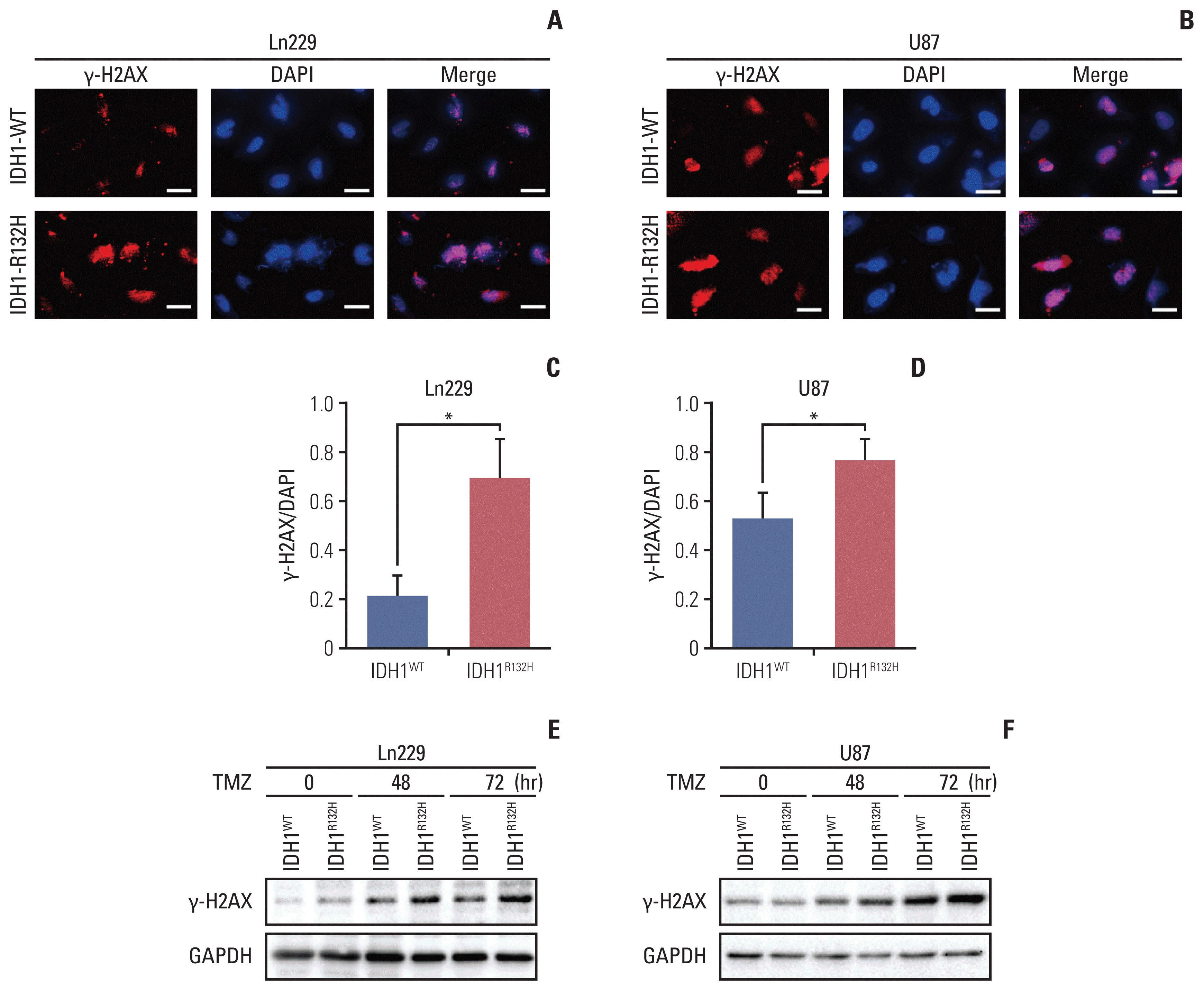 | Fig. 2Temozolomide (TMZ) induced more DNA damage in isocitrate dehydrogenase 1 (IDH1) mutation cells. (A, B) Immunofluorescence staining of γH2AX in glioma cells Ln229 and U87. Scale bars=100 μm. (C, D) Quantification of γH2AX is shown in A and B. *p < 0.05. (E, F) γH2AX expression in glioma cells Ln229 and U87 were detected by western blotting. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
|
3. IDH1 mutation enhanced TMZ induced apoptosis
We performed the annexin V/PI double staining to detect apoptosis. The result demonstrated that ATM inhibitor treatment enhanced the apoptosis induced by TMZ (Fig. 3A). Both annexinV+PI+ and annexinV+PI− labeled cells were more abundant in IDH1-R132H mutant cells (Fig. 3B and C). We found that the expression of apoptosis-related protein Bax was higher in IDH1-R132H mutant cells when treated with TMZ. The expression of anti-apoptotic protein Bcl-2 was reduced in IDH1 mutant cells compared to wild-type cells (Fig. 3D–F). The results demonstrated that TMZ induced more apoptosis in IDH1-R132H mutant glioma cells.
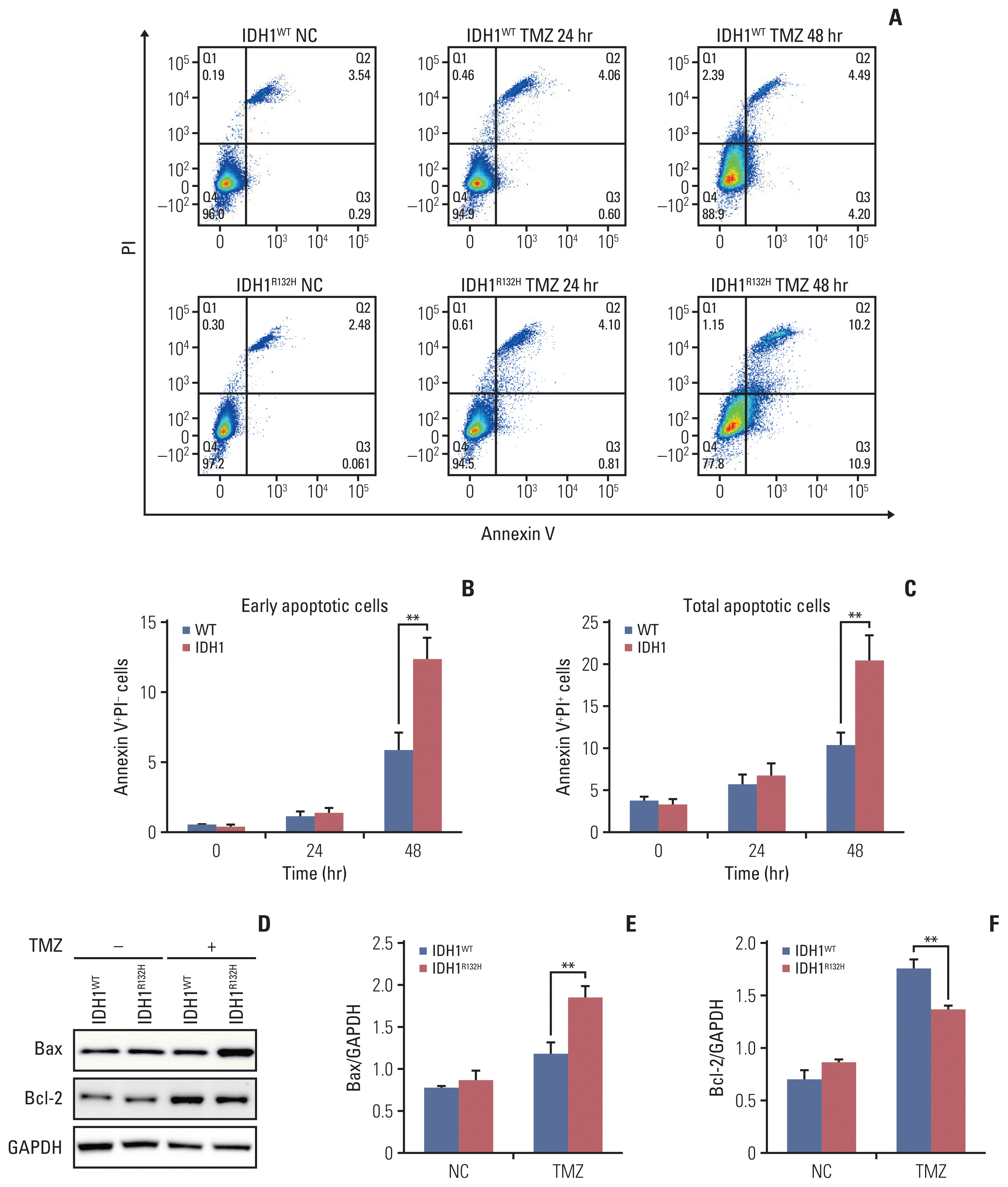 | Fig. 3Isocitrate dehydrogenase 1 (IDH1) mutant enhance temozolomide (TMZ) induced apoptosis. (A) Flow cytometry apoptosis analysis of IDH1 mutant and wild-type cells treated TMZ at different time points. (B) Statistical quantification of annexin V+propidium iodide (PI)− cells. (C) Statistical quantification of annexin V+PI+ cells. (D) Apoptosis-related protein Bax and Bcl-2 were detected by western blotting. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (E, F) Quantification of relative expression of Bax and Bcl-2. **p < 0.01.
|
4. IDH1 mutation inhibited ATM/CHK2 signaling triggered by TMZ
To further investigate the role of ATM signaling in TMZ induced DNA damage, the activation of ATM and its substrates CHK2 and p53 were detected. The western blot analyses showed significant phosphorylation of ATM, CHK2, and −p53 in both glioma cells when treated with TMZ (Fig 4A and B). We found that the expression of p-ATM was higher in wild-type cells compared with IDH1-R132H mutant groups. IDH1-R132H mutation also inhibited the activation of ATM downstream proteins including p-CHK2 and p-p53 (Fig. 4A and B). These data support that IDH1-R132H mutation inhibits ATM/CHK2/p53 signaling when treated with TMZ.
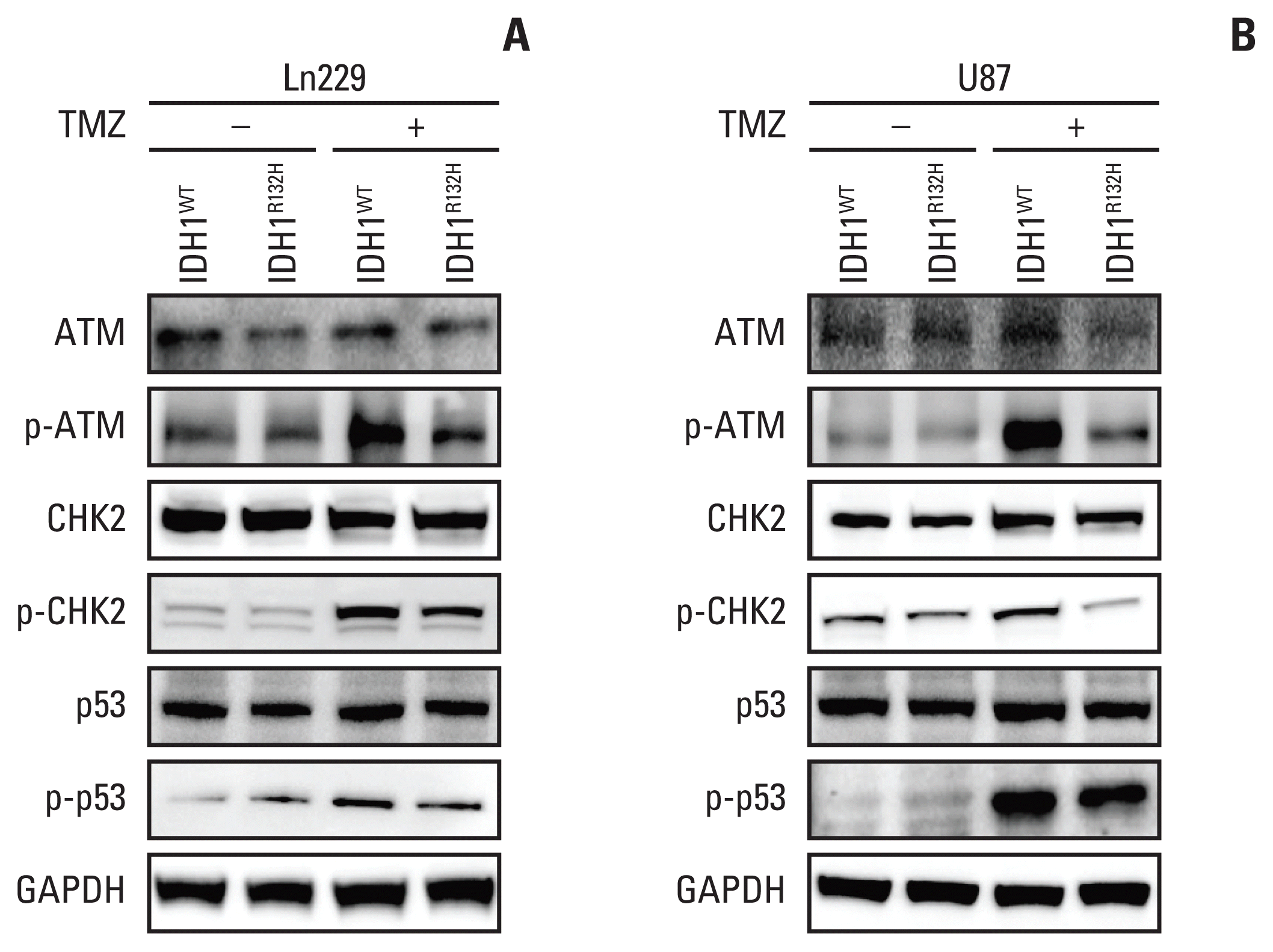 | Fig. 4Isocitrate dehydrogenase 1 (IDH1) mutation inhibits ataxia telangiectasia mutated (ATM)/checkpoint kinase 2 (CHK2) signaling triggered by temozolomide (TMZ). (A, B) The expression of ATM signaling including ATM, CHK2, p53, and their phosphorylated proteins in glioma cells treated with or without TMZ were detected by Western blotting. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
|
5. ATM inhibitor enhanced the antitumor effect of TMZ
Concerning IDH1-R132H mutation increased TMZ induced tumor damage via inhibiting ATM/CHK2/p53 signaling, we wondered if ATM inhibitor could strengthen the killing effect of TMZ. We added ATM inhibitor KU-55933 with or without TMZ to both wild-type and IDH1-R132H mutant groups. We found that ATM inhibitor combined with TMZ induced more apoptosis than TMZ alone group in IDH1-R132H mutant glioma cells. The antitumor effect of combination therapy was more significant than that of single-drug therapy in wild-type groups compared to IDH1 mutant groups. This may be IDH1 mutations inhibited the activation of ATM more significant than wild type. Moreover, whether the ATM inhibitor was added or not, TMZ induced more apoptosis in IDH1-R132H mutant cells than wild-type cells (Fig. 5A and B). Moreover, combination of ATM inhibitor and TMZ inhibited cell proliferation more significantly in IDH1 mutant glioma cells (Fig. 5C). Taken together, ATM inhibition enhanced the antitumor effect of TMZ.
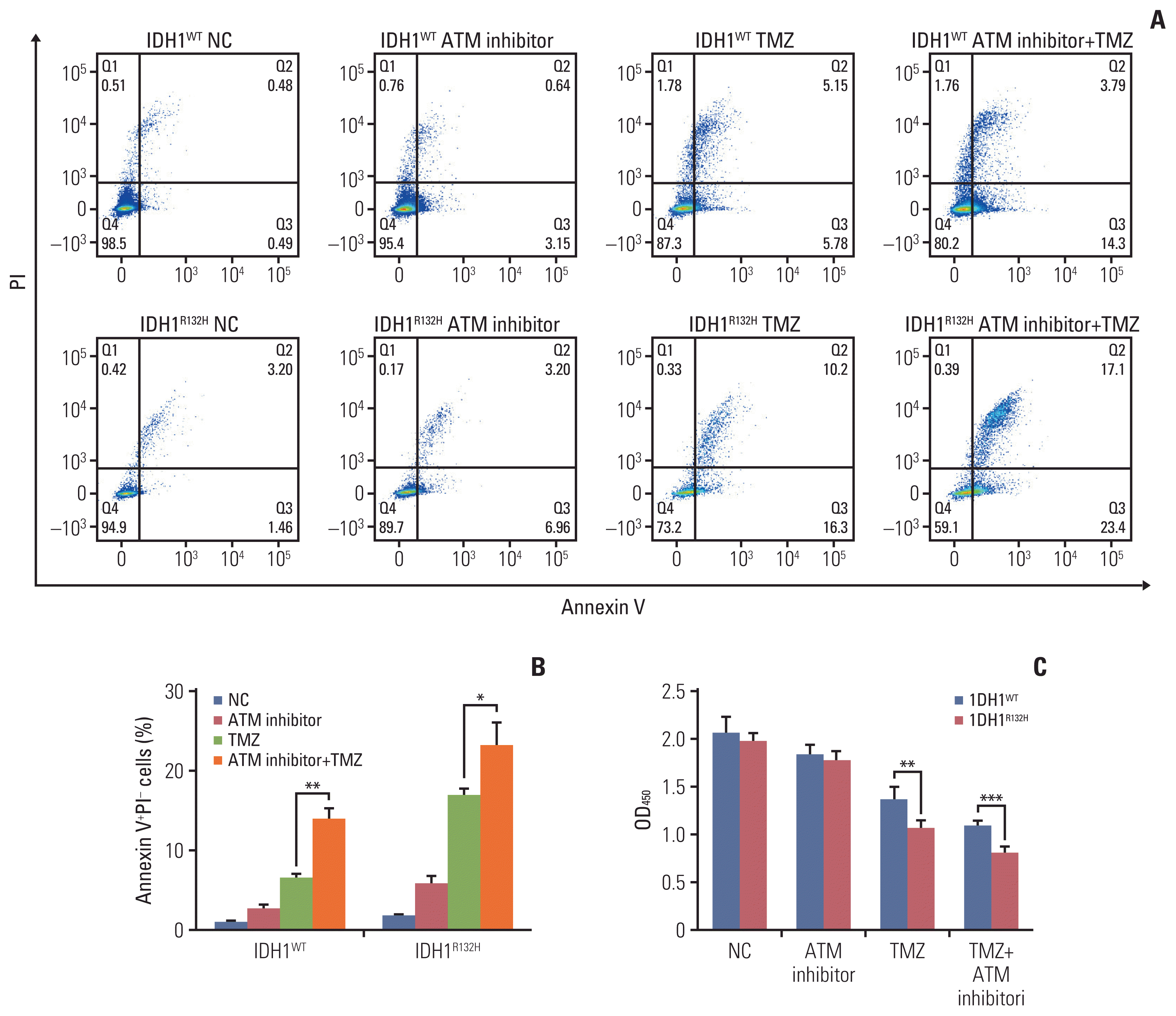 | Fig. 5Ataxia telangiectasia mutated (ATM) inhibitor enhanced the effect of temozolomide (TMZ) on tumor apoptosis. (A) Isocitrate dehydrogenase 1 (IDH1) mutation and wild-type gliomas were divided into four groups treated with negative control, ATM inhibitor, TMZ and ATM inhibitor+TMZ, after 72 hours, cell apoptosis was detected by flow cytometry. (B) Statistical quantification of annexin V+propidium iodide (PI)− cells. (C) IDH1 mutation and wild-type gliomas were treated with negative control, ATM inhibitor, TMZ and ATM inhibitor+TMZ, respectively. Cell proliferation was detected by Cell Counting Kit-8. *p < 0.05, **p < 0.01, ***p < 0.001.
|
6. Effect of IDH1 mutation on tumor in vivo
IDH1 mutant cells and wild-type glioma cells were subcutaneously injected into the back flanks of mice, and the tumorigenesis and tumor size were recorded. In the 5th week after subcutaneous inoculation of tumor cells, the same number of mice were killed, and the tumor size and weight were recorded (Fig. 6A). The results showed that the size and weight of IDH1 mutant gliomas were smaller than that of wild type (Fig. 6B and C). The results showed that IDH1 mutation had a certain inhibitory effect on tumor growth in vivo. The expression of Ki67 in IDH1 mutation group was lower than that in wild type (Fig. 6D). The results of immunohistochemistry showed that the score of wild-type Ki67 was significantly higher than that of IDH1 mutant group (Fig. 6E). In conclusion, IDH1 mutation can inhibit tumor growth in vivo.
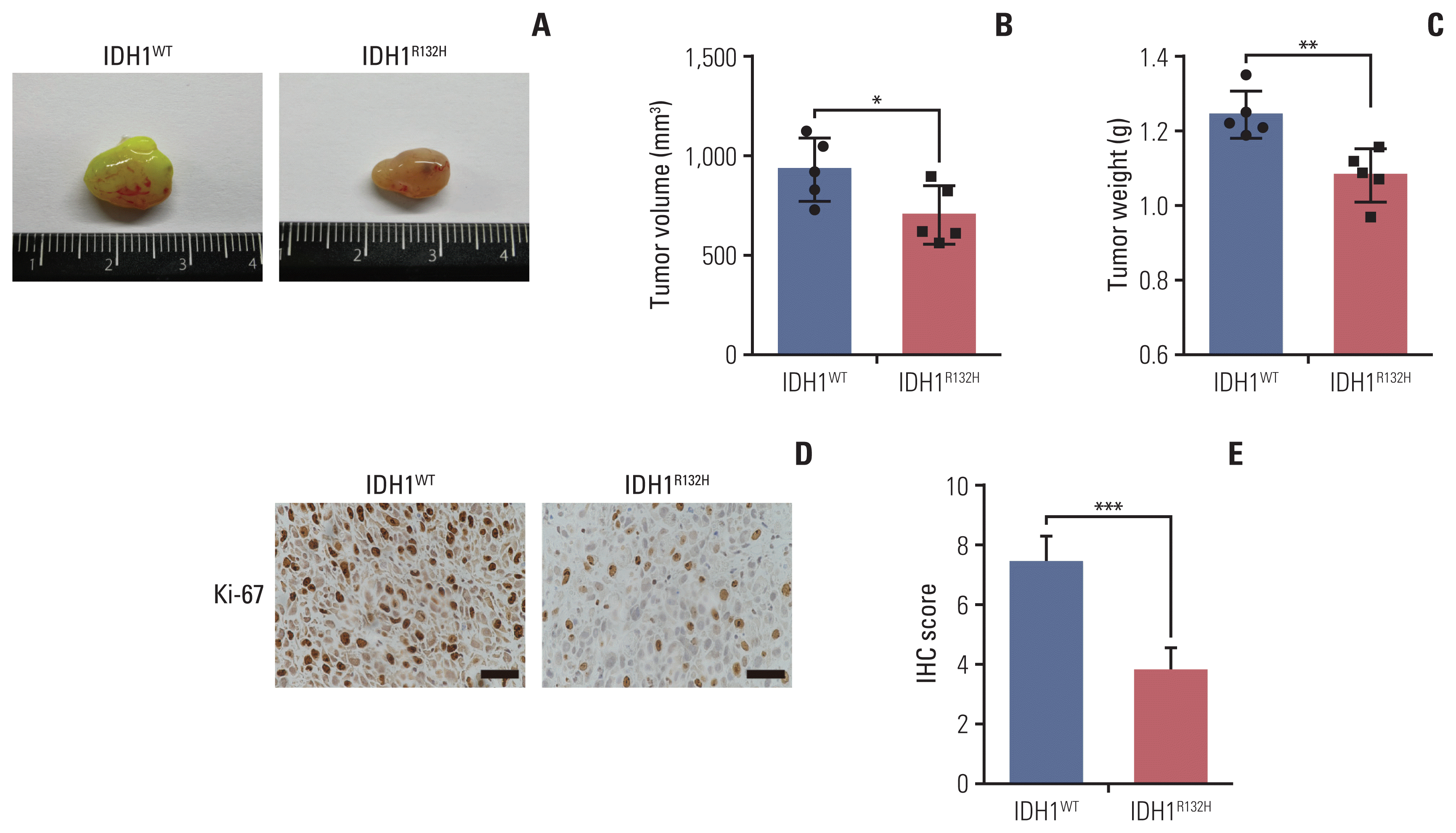 | Fig. 6The effect of isocitrate dehydrogenase 1 (IDH1) mutation on tumor in vivo. (A) The same number of IDH1 mutant and wild-type glioma cells were injected into the subcutaneous of 4–6 weeks nude mice, and the mice were killed and photographed in the fifth week. (B) The tumor size was detected and analyzed statistically. (C) The tumor weight was detected and analyzed statistically. (D) The tumor tissue was paraffin-embedded and stained with immunohistochemistry to detect the expression of Ki-67. Scale bars=25 μm. (E) The results of immunohistochemistry were scored and analyzed statistically. *p < 0.05, **p < 0.01, ***p < 0.001.
|
7. The effect of TMZ on IDH1 mutant tumor in vivo
To further study the effect of TMZ on IDH1 mutant glioma in vivo, we constructed subcutaneous tumor model in mice. Five weeks after tumor inoculation, TMZ was injected intraperitoneally for 7 days at a dose of 20 mg/kg. The tumor status was monitored, and the mice were killed at week seven. The size and weight of the subcutaneous tumor were recorded and analyzed statistically (Fig. 7A). We found that IDH1 mutant group was more sensitive to TMZ, and the tumor size and weight were significantly inhibited in this group (Fig. 7B and C). The expression of Bax was higher in IDH1 mutation group and Bcl-2 was lower in IDH1 mutation group (Fig. 7D and E). It is suggested that IDH1 mutation is more sensitive to TMZ, and chemotherapy might perform more antitumor effects.
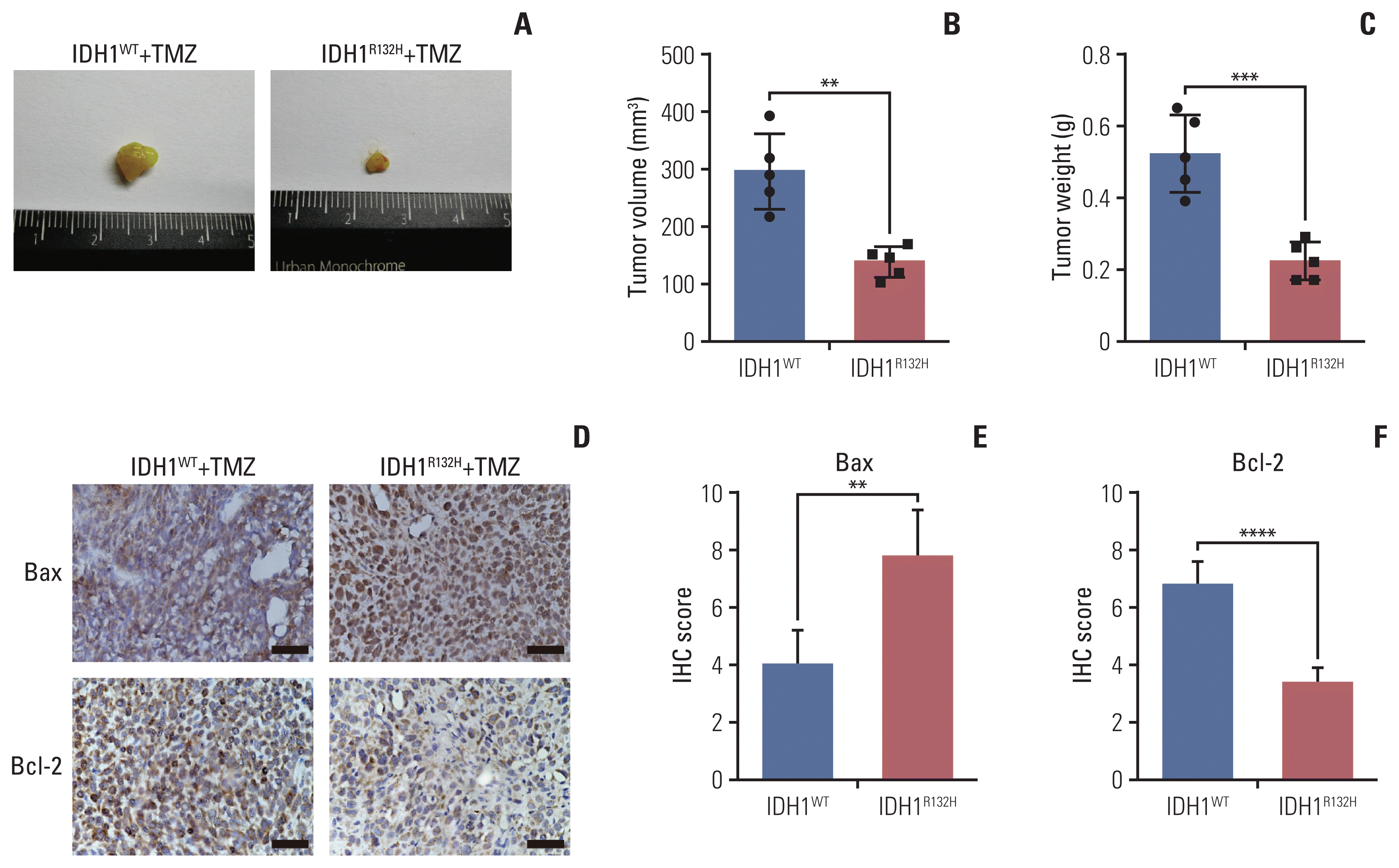 | Fig. 7The effect of temozolomide (TMZ) on isocitrate dehydrogenase 1 (IDH1) mutant glioma in vivo. (A) IDH1 mutant and wild-type glioma cells were subcutaneously injected into 4–6 weeks BALB/c nude mice. After transplanted for 4 weeks 20 mg/kg TMZ was administered intraperitoneally into mice for consecutive 7 days. All nude mice were killed in the sixth week. (B) The tumor volume was monitored and analyzed. (C) The tumor weight was monitored and analyzed. (D) Primary glioma sections from mouse models were stained with Bax and Bcl-2 for immunohistochemistry (IHC) assay. Scale bars=25 μm. (E, F) The results of IHC were scored and analyzed statistically. All results are expressed as the mean±standard deviation; n=5. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
|
Go to :
Discussion
The standard therapeutic schedule of glioblastoma is radiotherapy with daily TMZ. Though TMZ treatment prolonged the median survival time for about 2 months (from 12.1 months to 14.6 months), less than one-third of the patients survived for more than 2 years [13,14]. Genomic analysis showed that mutations of IDH1 accounted for a large proportion of gliomas. Researches demonstrate that about 60%–80% low-grade gliomas and most secondary glioblastomas have a mutation and most of those IDH1 mutations are replacing the arginine residue with histidine (IDH1-R132H) [15,16]. Tumors with IDH gene mutation have specific genetic and clinical characteristics, our previous studies identified IDH mutation with low ATRX expression were accompanied by longer progression time in glioma. To distinguish the IDH1-R132H mutation and ATRX expression status in primary and secondary gliomas is helpful for the choice of treatment strategy [17,18].
Studies of the role of IDH mutation on drug or radiation sensitivity were controversial. One kind of studies demonstrated that introduction of mutant IDH1 reduced TMZ resistance. IDH1 mutation impaired PARP1-mediated DNA repair signal in glioma [19]. Inoue et al. [20] showed that IDH1 mutation in acute myeloid leukemia impaired DNA repair and reduced hematopoietic stem cells. Patients with oligodendroglioma tumors that occurred IDH mutation benefit from alkylating-agent chemotherapy with radiotherapy [21]. However, some studies also demonstrated that mutant IDH1 expression increased TMZ resistance due to enhance the RAD51-mediated homologous recombination [9]. A clinical research also suggested that IDH1 mutation conferred resistance to TMZ. They indicated that there were no differences in overall survival between the TMZ plus radiotherapy or radiotherapy group [22]. Though the prognostic value for IDH mutation in clinic has been observed, the role of IDH1 mutation on drug sensitivity is still under debate. Therefore, the molecular characteristics of IDH1 mutant cells in response to TMZ need to be further investigated and the development of more effective treatment is still an important priority for patients with glioma.
As a DNA-methylating agent, TMZ has antitumor effect in the treatment of malignant glioma. TMZ induced multiple DNA adducts, of which the O6 methylguanine was more cytotoxic [23]. O6 methylguanine mispairs with thymine and is recognized by the mismatch repair. In the process of this error repair, secondary damage is formed, which blocks DNA replication in replication cycle, resulting in DNA DSBs [24]. DSBs finally trigger apoptosis, necrosis, and autophagy. DNA damage repair involves a protein network, which can make appropriate sense and response to DNA damage, and the interference of this network can promote the occurrence of tumor. Activated ATM recruits DNA repair mechanisms, activates cell cycle checkpoints, and induces apoptosis by phosphorylating CHK2 and tumor suppressor p53 [25,26]. It is suggested that IDH1 has a TET2 independent effect on DNA repair in acute myeloid leukemia cells, and its mechanism may be related to the down-regulation of ATM-mediated by histone methylation [20]. Whether TMZ mediated DNA damage affects ATM signal pathway in IDH1 mutation of glioma was under debate.
Our data suggest that IDH1-R132H mutant gliomas are sensitive to chemotherapy which correspond to the clinical observation in the previous research. Compared with the wild-type group, the glioma cell proliferation in the IDH1 mutant group was significantly inhibited by TMZ. A lower dose of TMZ was effective to induce cytotoxic damage in IDH1-R132H mutant cells. The IC50 of TMZ was significantly decreased in IDH1-R132H mutant gliomas, for U87 cells, IC50 of IDH1 WT cells was 543 μmol/L while IC50 of IDH1-R132H cells was 259.7 μmol/L. Consistent with previous findings [19], TMZ induced more DNA damage in IDH1-R132H mutant cells due to the high expression of γH2AX. We confirmed that IDH1 mutations elevated cell apoptosis induced by TMZ, as Annexin V–positive cells were increased in IDH1-R132H mutant group. Interestingly, the baseline apoptosis was also elevated in mutant group. The TMZ induced activation of ATM and its substrates CHK2 and p53 were inhibited in IDH1-R132H mutation group. Due to these molecular mechanisms, IDH1 mutant gliomas are sensitive to TMZ therapy. Furthermore, ATM inhibitor enhanced the cytotoxic effects of chemotherapy in IDH1-R132H glioma cells.
Taken together, our findings demonstrate that IDH1 mutations enhance TMZ sensitivity via regulating ATM/CHK2 pathway in glioma. We provided the preclinical evidence that combination of TMZ and ATM inhibitor enhanced the antitumor effect in IDH1 mutant gliomas. TMZ treatment combined with DNA repair inhibitor might be served as a prospective therapeutic strategy for glioma.
Go to :
 XML Download
XML Download