

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Normal coagulation is a balance between hemostatic and fibrinolytic processes, the loss of which may result in either excessive bleeding or intravascular thrombosis, which defines coagulopathy. Abnormalities in coagulation testing may also be considered evidence of coagulopathy, even in the absence of clinical sequelae of bleeding or thrombosis.
There is limited data on the incidence of coagulopathy in neurocritical care units; however, it is commonly seen in critically ill patients with incidences ranging widely from 14% to 81% [1,2]. Coagulopathies may be acquired through a variety of conditions including trauma, organ failure and the use of medications, and may confer an increased risk of secondary hematoma expansion, poor functional outcome, and death across the spectrum of neurocritical illnesses [3-6]. An aging patient population and increased use of antithrombotic and anticoagulant agents demand unique considerations [3,7,8]. Timely, appropriate assessment and treatment are warranted to mitigate hematoma expansion and to facilitate emergent neurosurgical intervention when indicated. We discuss herein techniques in the assessment and management of coagulopathies for the patient with an intracranial hemorrhage, in line with the most recent guidelines adopted by the American Heart Association (AHA), American Stroke Association (ASA), Neurocritical Care Society (NCS), and Society of Critical Care Medicine (SCCM) [3,9]. We include updates from clinical trial findings that were not available at the time of these publications.
Go to : 
COAGULATION ASSESSMENT
Conventional coagulation tests
Common (or conventional) coagulation tests (CCT) include prothrombin time/international normalized ratio (PT/INR), activated partial thromboplastin time (aPTT), platelet count, D-dimer, and fibrinogen levels. The PT is a laboratory test developed to assess the function of the “extrinsic pathway” whereby calcium and tissue factor (TF) are added to citrated blood and the time to coagulation is measured. To correct for interlaboratory differences in TF preparations, the INR was developed and is only intended for monitoring the effect of warfarin therapy [10]. Abnormalities in PT may reflect coagulopathy seen in liver failure, disseminated intravascular coagulopathy (DIC), trauma as well as in the case of some medications such as factor Xa inhibitors. The aPTT was developed historically to assist in the diagnostic process for patients who exhibit signs consistent with hemophilia [11]. In modern clinical practice, it is commonly used to assess the “intrinsic pathway” of hemostasis and is performed by adding calcium, phospholipid, and an activator such as kaolin to citrated blood, and the time to coagulation is measured. It is most useful for monitoring the effect of unfractionated heparin (UFH); however, it cannot reliably reflect the effect of other anticoagulants [12].
CCTs present limitations in our patient population as they are plasma-based, and hence cannot measure interactions between clotting factors, TF, and platelets, and were not designed to assess hemostatic integrity in the trauma or preoperative patient [11,13]; they simply reflect a static evaluation of the coagulation cascade with clot formation as their endpoint rather than assessing the whole coagulation system [13]. They have also been shown to correlate poorly with clinical bleeding and transfusion requirements, lack accuracy in detecting deficiencies in coagulation factors, fail to detect the effects of novel anticoagulation agents or antiplatelet therapy (APT), and do not describe platelet function and fibrinolysis.
Viscoelastic hemostatic assays
Shortcomings of CCTs have led to increased utilization of viscoelastic hemostatic assays (VHAs), which offer a better depiction of the successive steps that comprise the cell-based theory of hemostasis, i.e., initiation, amplification, propagation, and termination through fibrinolysis. VHAs are performed by placing whole blood in a cup with a suspended pin, which transduces changes in tension during clot formation and breakdown with rotation (Fig. 1) [14].
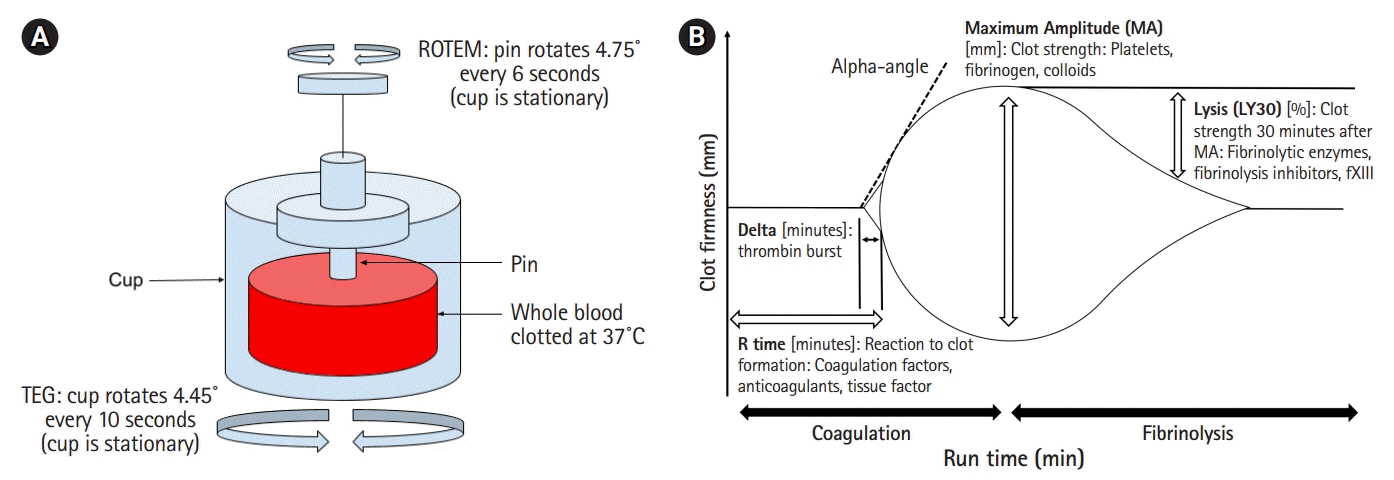 | Fig. 1.(A) Schematic of viscoelastic testing with specimen of whole blood in cup. With thrombelastography (TEG) the cup oscillates with pin remaining stationary, while with ROTEM pin oscillates while the cup remains stationary. Measurement of pin synchronization with the cup reflects the stages of clot formation. (B) TEG recording with measurement parameters.
|
Thrombelastography (TEG) is a VHA commonly used in North America. TEG measures different phases of the coagulation cascade including time to initiate clot formation (reaction time [R]), rate of clot formation (kinetics [K], α angle), maximum clot strength (maximum amplitude [MA]), and clot stability (fibrinolysis; Ly30). When compared with CCT, TEG has been shown to be a better predictor of significant bleeding, the need for massive transfusions as well as mortality at 24 hours and 30 days following trauma [15]. Furthermore, there is a reported mortality benefit to TEG-directed hemostatic resuscitation in trauma patients requiring massive transfusions when compared to interventions dictated by CCTs [16-18]. An example of CCT and TEG guided transfusion recommendations implemented at our institution is shown in Table 1.
Table 1.
Memorial Hermann Hospital transfusion recommendations based on abnormal coagulation testing in bleeding patients
Adapted from Holcomb et al. [15], with permission of Wolters Kluwer Health.
ACT, activated clotting time; FFP, fresh frozen plasma; RBC, red blood cells; MA, maximum amplitude; Ly30, percentage of lysis 30 minutes after MA; PT, prothrombin time; aPTT, activated partial thromboplastin time; INR, international normalized ratio.
![]()
Platelet function
While CCT identifies patients at increased risk of bleeding due to thrombocytopenia, it does not indicate; however, qualitative platelet dysfunction due to the use of APT, renal insufficiency or other factors. Bleeding time has grown out of favor for this purpose due its operator-dependence and lack of sensitivity [19]. Light transmission aggregometry in platelet-rich plasma or whole blood is considered the gold standard for assessment of platelet function, but availability may be limited due to poor standardization and time consumption [20]. Commercially available point-of-care platelet function assays overcome some of these obstacles and include the platelet function analyzer (PFA-100, Siemens Medical Solutions, Malvern, PA, USA), VerifyNow (Accumetrics, San Diego, CA, USA) ASA and P2Y12 assays. These tests detect dysfunction secondary to the use of antiplatelets and can provide measures of patient response to these agents as well as adequacy of efforts to reverse them. Standard TEG testing unreliably detects the presence of single APT use; however, it may detect coagulopathy seen in combination APT use [21]. TEG with platelet mapping (TEG-PM, Haemoscope Corp., Niles, IL, USA) is a specific VHA that has been shown to correlate with platelet aggregometry and is able to detect platelet dysfunction due to APT and other coagulopathies. In neurocritically ill patients, such as those with subarachnoid hemorrhage (SAH), intracerebral hemorrhage (ICH), or traumatic brain injury (TBI), platelet dysfunction may be seen even in the absence of APT use or failure of other organ systems and may confer worse outcomes [22-24].
Go to :
COAGULOPATHY IN ACUTE BRAIN INJURY
Historically, reports dating back to the 1970s described baseline hypercoagulability based on TEG in patients with acute ischemic stroke (AIS) and SAH [25,26]. The more recent use of TEG in the neurocritical care unit has allowed investigators to better elucidate coagulopathies seen in different types of acute brain injury. In general, patients with acute brain injuries present with hypercoagulable states compared with normal controls. The impact of this response, variations in this response between patients with the same type of injury, and potential therapeutic implications largely remain unclear.
Acute ischemic stroke
Patients with AIS have been reported to present with a hypercoagulable state when compared with normal controls. In a study of patients with AIS presenting within the window of administering tissue plasminogen activator (tPA) (i.e., within 3 hours), TEG demonstrated a hypercoagulable state based on shorter R and K times with greater α angle. After treatment with tPA, significant changes in MA, G, and Ly30 were demonstrated within 10 minutes [27]. However, the same group was unable to predict a clinical response to tPA treatment using TEG values [28].
Admission TEG values have been associated with outcomes after AIS. In another study utilizing TEG in patients with AIS, MA was found to be an independent predictor of poor outcome (modified Rankin Score ≥2) at 1 year. Recurrence rate of ischemic events were found to be higher in the 3rd MA tertile group, despite higher rates of treatment with dual APT. Higher tertile of MA was also associated with stroke severity (higher National Institutes of Health Stroke Scale scores on admission and longer hospital stay) [29]. Although MA appears to be a marker of stroke severity and may portend worse outcome, it is unclear if this may be a potential target for intervention.
Intracerebral hemorrhage
Studies utilizing TEG in patients with ICH have illustrated its potential in predicting the clinical course. In a study by Kawano-Castillo et al. [30], patients with ICH demonstrated faster and stronger clot formation at baseline (shorter R and delta) and stronger clot strength (higher MA and G) at 36 hours compared with normal controls. Patients with hematoma expansion had significantly longer baseline K and delta compared with nonexpanders, indicating that the former group had slower clot formation [30]. Other reports have also demonstrated greater baseline hypercoagulability in patients with ICH [31,32].
Although patients with ICH often present with a hypercoagulable state compared with controls, hypocoagulability may have a powerful impact on outcome from ICH. Hypocoagulability by TEG has been associated with worse functional outcome, higher rates of cerebral herniation, and mortality from ICH and isolated TBI [33]. Detection of hypocoagulability could represent an application of TEG in terms of therapeutic implications in ICH.
Subarachnoid hemorrhage
Like AIS and ICH, baseline coagulation disturbances are common in the spontaneous SAH population. In animal models of SAH, hypercoagulability is demonstrated as early as 30 minutes following the injury [34]. In patients with SAH, early hypercoagulability and platelet dysfunction has been identified by TEG and shown to correlate with poor outcomes, including an increased incidence of delayed cerebral ischemia and worse modified Rankin Scores at 3 months [22]. Similar to AIS, there is an association noted between elevated MA values and poor outcomes, independent of other inflammatory biomarkers, age, and Hunt-Hess grade [35].
Traumatic brain injury
Trauma population studies have elucidated some of the complex coagulation disturbances which occur in the setting of traumatic injury. Furthermore, more widespread use of TEG has enhanced the ability to detect coagulation differences. In general, hypercoagulability by TEG is the most commonly observed pattern in trauma and may be observed in up to 65% of patients [36]. However, a subset of patients may present with an acute traumatic coagulopathy. Hypocoagulability on admission is associated with a several-fold increase in morbidity and mortality [37]. Findings seen most often with severe injuries include elevated PT/partial thromboplastin time, low platelet counts, and fibrinogen levels on CCT. TEG has demonstrated hypocoagulability in severe trauma through changes in the values of R, α angle, and MA. Hyperfibrinolysis, while only seen in 2% to 6% of patients, may have a powerful impact on outcome from major trauma. Ly30 values greater than 3% have been associated a two-fold increase in mortality in trauma patients [15]. Patients with massive tissue injury have also demonstrated evidence of hyperfibrinolysis, such as elevated D-dimer and fibrin degradation products on CCT [38]. As previously discussed, TEG based protocols may now be used to guide transfusion therapy in the setting of traumatic injury.
Coagulopathy following TBI is associated with the severity of injury. In one report, it was present in up to one-third of patients with isolated TBI and up to 60% of patients with severe TBI, although there have been reports of lower rates [39]. The brain is rich in TF and it is postulated that its release activates the extrinsic pathway, which in turn leads to a consumptive coagulopathy and hyperfibrinolysis. Evidence of coagulopathy on TEG (e.g., increased R or decreased MA) following isolated TBI has been shown to be associated with considerably higher rates of mortality when compared to TBI patients without evidence of coagulopathy (66% vs. 16.6%) [40]. Hyperfibrinolysis also continues to play an important role in predicting poor outcome in patients with TBI, with D-dimer at admission shown to be an independent risk factor for poor outcome [41].
Go to :
COAGULOPATHY IN SYSTEMIC DISEASE
Acute liver failure
Acute liver failure is associated with a deficiency of both procoagulant and anticoagulant proteins. While CCT may show an elevated PT/INR in this patient population, TEG has demonstrated that the vast majority of these patients have a normal ability to form clots and may even be hypercoagulable [42].
Thrombocytopenia
Thrombocytopenia may be seen in a variety of conditions encountered in the neurocritical care unit, including sepsis (which is the most common cause of thrombocytopenia in critically ill patients), hypersplenism, DIC, blood loss, mechanical fragmentation, medications, bone marrow suppression, and immune-mediated disorders [43]. In a case series by Chan et al. [44] of patients with thrombocytopenia undergoing neurosurgical procedures, a platelet count <100,000/µL was associated with a significant increase in the rate of postoperative hematoma formation when compared with patients with a platelet count >100,000/µL. Current guidelines recommend a transfusion threshold of <100,000/µL for patients with intracranial bleeding or those undergoing a neurosurgical procedure.
Disseminated intravascular coagulopathy
DIC is associated with multiple disease entities encountered in critically ill patients, most commonly due to sepsis, although trauma and malignancy are also common causes encountered in the neurocritical care unit [45]. It is characterized by widespread microvascular thrombosis due to TF expression, leading to massive fibrin deposition, consumption of platelets and coagulation factors, hyperfibrinolysis, hemorrhage, and organ failure [46]. The management of DIC is that of the underlying condition and CCT-guided transfusion if a patient is actively bleeding or at high risk of bleeding.
Uremia
Uremia is associated with an increased risk of hemorrhage secondary to platelet dysfunction. A rising prevalence of chronic kidney disease and the use of renal replacement therapy has led to this being encountered with increasing frequency in the neurocritical care unit as a cause of, or contributing to, intracranial bleeding [47,48]. Desmopressin (DDAVP) is the agent most commonly used in the treatment of uremic bleeding and has been shown to reduce bleeding time and normalize hemostasis in patients with uremic platelets undergoing surgery [49,50]. Its actions are mediated by an increase in endothelial release of von Willebrand factor and platelet membrane glycoprotein expression, which in turn promotes platelet adhesion to the endothelium [51,52]. It has been shown to restore platelet function within 30 minutes of administration, though its effects are short-lived at around 3 hours [53]. DDAVP is dosed at 0.4 µg/kg administered intravenously for this indication and is well tolerated with few reported side effects [3,12,54].
Go to :
REVERSAL OF ANTITHROMBOTIC THERAPY
There has been a significant, continued increase in the use of antithrombotic agents among patients admitted to the neurocritical care unit, due to an aging population and increased diagnosis of ischemic events that warrant such therapies. The introduction of novel agents poses a diagnostic and therapeutic dilemma for the neurointensivist when managing the patient with intracranial hemorrhage. Antithrombotic agents may be roughly divided into APT and anticoagulant therapy.
Antiplatelet therapy
The main classes of antiplatelet drugs commonly used in practice include cyclooxygenase-1 (COX-1) inhibitors (e.g., acetylsalicylic acid; ASA or aspirin), phosphodiesterase inhibitors (e.g., dipyridamole and cilostazol), P2Y12 receptor inhibitors (e.g., clopidogrel, prasugrel, and ticagrelor) and glycoprotein IIb/IIIa inhibitors (e.g., abciximab, eptifibatide, and tirofiban). Aspirin use confers an absolute risk increase of 0.1% per year of ICH compared to control [55]. Clopidogrel carried a similar rate of ICH incidence when compared to aspirin in the Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial [56]. Prasugrel, when compared to clopidogrel in the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction 38 (TRITON-TIMI 38) trial, demonstrated a higher risk of life-threatening bleeding overall although the rate of ICH was similar between the two groups [57]. Ticagrelor was associated with an increased rate of intracranial hemorrhage when compared to clopidogrel in the Platelet Inhibition and Patient Outcomes (PLATO) trial, although this difference was nonsignificant [58]. Single APT use is associated with a low risk of major bleeding, although this risk increases significantly when combination APT are implemented, similar to the risk of anticoagulants [12,59]. In a study of a large cohort of patients from the Get With The Guidelines-Stroke (GWTG-Stroke) database with ICH, the use of combination APT, but not single APT, was associated with a higher risk for in-hospital mortality when compared with patients not taking APT [60].
Reversal of APT in the bleeding patient may be achieved through the administration of platelet transfusion. Routine platelet transfusion for ICH based on reported use of APT alone was associated with an increased rate of poor functional outcome in the Platelet Transfusion in Spontaneous Intracerebral Hemorrhage (PATCH) trial [61]. It is important to note that this trial did not utilize qualitative platelet function testing and excluded surgical patients, so platelet transfusion may be appropriate for patients who require neurosurgical intervention. In a study by Choi et al. [62] of 107 patients presenting with traumatic intracranial hemorrhage and reported APT use, a significant percentage of patients had subtherapeutic ASA/P2Y12 assays that would be unlikely to benefit from platelet transfusion. Among patients that did receive platelet transfusions, the amount transfused did not adequately reverse its effect in almost half of this patient cohort [62]. There may, therefore, be a role for targeted platelet transfusions based on quantitative assessment of platelet function before and after transfusion. This was illustrated in a retrospective study by Naidech et al. [63] on a series of patients with ICH and abnormal platelet function activity. Early (<12 hours from symptom onset) platelet transfusion improved platelet activity assay results and was associated with smaller final hemorrhage size and more independence at 3 months (modified Rankin Score <4) [63]. Similarly, in a TBI population, TEG-directed platelet transfusion was associated with a decreased mortality compared to a historical cohort in a retrospective study by Furay et al. [64].
DDAVP use has also been demonstrated to improve platelet function in patients on COX-1 and ADP receptor inhibitors in several tests of platelet function compared to those who had not received reversal agents [3,50,51,65-68]. Clinically, it has been shown to reduce blood loss and improve hemostasis in patients with aspirin exposure undergoing cardiac surgery [49,69,70]. In two small studies of patients with intracranial hemorrhage and either reduced platelet activity on PFA-100 and/or known aspirin use, DDAVP administration was associated with restoration of platelet function on repeat testing [53,71]. Given its low cost and relatively good safety profile, its administration should be considered in patients with intracranial hemorrhage who were exposed to antiplatelet agents [3].
Anticoagulation therapy
The main classes of anticoagulant drugs include vitamin K-dependent coagulation factor antagonists (VKA, e.g., warfarin), factor Xa inhibitors (e.g., fondaparinux, rivaroxaban, apixaban), direct thrombin inhibitors (DTI, e.g., argatroban, bivalirudin, dabigatran), and heparinoids (i.e., unfractionated, or UFH, and low-molecular-weight heparin [LMWH]). In the neurocritical care unit, common indications for these medications include stroke prevention in atrial fibrillation and treatment of venous thromboembolism. Reversal strategies for anticoagulation-associated intracranial hemorrhage are summarized in Table 2.
Table 2.
Summary of common anticoagulation agents and Neurocritical Care Society guidelines for the reversal in intracranial hemorrhage
Adapted from Frontera et al. [3], with permission of Springer Nature.
IV, intravenous; PCC, prothrombin complex concentrate; FFP, fresh frozen plasma; CrCl, creatinine clearance.
![]()
Vitamin K antagonists
VKA inhibit vitamin K-dependent factors in the coagulation cascade: factors II, VII, IX, and X. VKA activity can be assessed by PT/INR. The risk of bleeding while taking a VKA increases with the duration of therapy and higher INR levels. For each increment in INR elevation above the therapeutic range, the risk of bleeding on a VKA doubles [72].
Vitamin K replacement is essential to replenish the vitamin K-dependent factors and reverse VKA activity. It should be given promptly and IV administration mitigates variability in oral vitamin K absorption. Although the risk profile is low, it may take 24 hours or more to become effective [3].
Fresh frozen plasma (FFP) has been conventionally used to reverse VKA in conjunction with vitamin K. Although it is relatively inexpensive and widely available, its use is complicated by delays in administration, potential transfusion related reactions, and large volumes which may be required for full INR reversal. This had led to the more widespread use of prothrombin complex concentrate (PCC). PCC is derived from plasma and contains factors II, VII, IX, and X in variable proportions in different preparations. It is rapidly administered in a small volume. PCC has been demonstrated to reverse INR to <1.4 and maintain INR reversal for >48 hours in the majority of patients on VKA therapy [73]. In the INR Normalization in Coumadin Associated Intracerebral Haemorrhage (INCH) trial, a randomized trial comparing FFP and PCC for VKA reversal in intracranial hemorrhage, PCC provided more rapid INR reversal with an effective reduction in hematoma expansion [74]. The trial was stopped early due to safety concerns with FFP therapy. Both the AHA/ASA as well as the NCS/SCCM guidelines currently recommend consideration of PCC over FFP for VKA reversal in ICH [3,9].
Factor Xa inhibitors
Xa inhibitors prevent the conversion of prothrombin to thrombin. Detection of novel agents using calibrated chromogenic assays are expensive and not readily available, and the utility of existing anti-Xa assays have not been validated to identify the presence of the oral factor Xa inhibitors. There is data to support the use of TEG to detect coagulopathies induced by these agents, which may inform reversal strategies in these patients [75].
PCC is commonly used for Xa inhibitor reversal. PCC has been demonstrated to effectively reverse rivaroxaban in healthy volunteers [76]. In the setting of intracranial bleeding, it is recommended to administer 50 units/kg if the medication was ingested within 3 to 5 half lives or if the time of last exposure is unknown. In patients with a known ingestion within 2 hours, activated charcoal may also be utilized if this is deemed safe from an airway protection standpoint. Hemodialysis is not effective in removing Xa inhibitors [3].
Andexanet alpha is a recombinant inactive form of factor Xa. It binds to Xa inhibitors with high affinity and sequesters the medication, resulting in reduced anti Xa activity. In the Andexanet Alfa, a Novel Antidote to the Anticoagulation Effects of FXA Inhibitors-4 (ANNEXA-4) study of 352 patients with major bleeding, andexanet alpha reduced anti Xa levels by 92% in rivaroxaban/apixaban treated patients and 75% in enoxaparin [77]. Andexanet alpha is approved by the U.S. Food and Drug Adminstration for the reversal of rivaroxaban and apixaban, but widespread use is limited due to the cost of the medication.
Direct thrombin inhibitors
DTI directly inhibit the activity of factor IIa, which is the key factor in converting fibrinogen to fibrin. DTI have an additional unique indication in the treatment of heparin induced thrombocytopenia. Intravenous formulations are short acting and generally do not require reversal agents. However, the reversal of oral dabigatran was challenging before idarucizumab became available. Idarucizumab is a monoclonal antibody which binds to dabigatran with considerably higher affinity than factor IIa. In the Reversal Effects of Idarucizumab on Active Dabigatran (RE-VERSE AD) study of 503 patients with life-threatening bleeding (Group A) or need for emergent surgical procedure (Group B), idarucizumab reversed thrombin time to normal in 100% of patients and this effect remained relatively stable 24 hours after treatment. Normal intraoperative hemostasis was achieved in 92% of the patients in Group B [78]. The rate of thrombotic events in this study was similar to those reported after major surgical procedures or hospitalization for uncontrolled bleeding, and may be attributable to the low rate of reinitiation of anticoagulation [79,80]. Idarucizumab is administered in 2 doses of 2.5 g given within 15 minutes. Activated charcoal is an additional option for dabigatran, similar to Xa inhibitors, and is recommended for consideration in the AHA/ASA as well as the NCS/SCCM guidelines [3,9].
If idarucizumab is not available, hemodialysis and PCC are alternative options. Dabigatran is renally excreted and is effectively removed by hemodialysis. However, there is a theoretical risk of worsening cerebral edema in patients with mass lesions. The recommended dosing for PCC is 50 units/kg, the same as for Xa inhibitors. Administration of PCC beyond 3 to 5 half lives of dabigatran exposure may be considered in patients with renal insufficiency [3,9].
Heparin and low-molecular-weight-heparin
UFH activates antithrombin III activity, which inhibits factors IIa, and Xa. Heparin activity can be assessed utilizing aPTT or point-of-care activated clotting time. Heparin activity may also be assessed with TEG; shortening of R time with heparinase implicates the presence of heparin in the sample as the cause of coagulopathy. Its effects may be reversed with protamine, which is a naturally occurring protein that binds to heparin and its dosing is outlined in Table 2.
LMWH has a similar mechanism of action but is longer acting and thought to have more predictable pharmacology in the setting of normal renal function. Assessment of LMWH activity requires the use of an anti-FXa assay. Protamine may be used to reverse LMWH activity, but this reversal is incomplete and estimated to be around 60% [3]. A novel approach to LMWH is andexanet alpha. Although this is a potential therapeutic intervention with a possibility of more complete LMWH reversal than with protamine, it is currently not approved for this indication and is still undergoing further study [77].
Go to :
CONCLUSION
Coagulopathy is commonly encountered in the neurocritical care unit and poses a challenge to the clinician when managing the patient with intracranial hemorrhage. Utilization of viscoelastic testing has shown great promise in this arena, allowing one to risk stratify patients and guide transfusion requirements. As the widespread use of antithrombotic therapy continues to increase, further development of specific testing for individual medications and targeted reversal agents would improve the management of hemorrhagic complications.
Go to :
 XML Download
XML Download