

 PDF
PDF Citation
Citation Print
Print


Introduction
Targeted therapeutics against kinase families and membrane proteins have dramatically improved the outcome of cancer patients using their target biomarkers [1]. A common strategy to identify biomarkers for cancer therapy is to compare various profiles between cancer and normal samples. So, a minimum number of samples are required for statistical significance. However, recently, important targets found in very few cases have been identified. Furthermore, it is difficult to discover extremely overexpressed genes with rare events using standard statistical methods including the t test [2]. The traditional statistical strategies may not reflect the clinical and molecular heterogeneity of cancer. To overcome this limitation, outlier analysis methods such as ’cancer outlier profile analysis (COPA)’ have been developed to discover genes with genetic heterogeneity, such as up-regulation of oncogenes via chromosomal translocation, in a subset of cancer samples [3]. Several oncogenes including TMPRSS2-ETS and SPINK in prostate cancer and ALK, FGFR2, KIT, NTRK1, NTRK2, PDGFRA, and RET in colorectal cancer cell lines have been identified as outlier genes using this approach [2,4]. In fact, some current promising targets such as RET and ROS1 are present in very small numbers (less than 5%, sometimes less than 1%) in lung cancer populations [5].
Based on extremely overexpressed targets with very low prevalence, we hypothesized that genes showing outlier patterns with rare events among cancer samples can be oncogenic drivers and therapeutic targets for precision medicine. The COPA method uses the median and median average difference (MAD) to detect outliers in a subset of cases [3]. Tukey’s fences method is also one of the several methods available for detecting outliers in an individual sample. This method is also widely used to display boxplots using the median, quartile, and extreme values of a dataset. Even though Tukey’s fences method is advantageous for analyzing data that do not follow a normal distribution because it does not depend on a mean or standard deviation, this method may not be suitable for small dataset analysis [6]. Here, we applied not only a modified Z-score, like COPA, but also a modified Tukey’s fences outlier analysis to discover extreme outlier genes as therapeutic cancer targets.
We analyzed the mRNA expression profiles of the Cancer Cell Line Encyclopedia (CCLE) and The Cancer Genome Atlas (TCGA) datasets. The analyzed genes were classified into three groups—kinase group (KG), membrane group (MG), and other group (OG)—based on The Human Protein Atlas (THPA) database. We also analyzed the mechanism underlying the high expression of outlier genes, the DNA copy number, and DNA methylation status of each gene between samples with outliers and others using TCGA datasets. Two genes, TM4SF4 and LRRK2, were chosen as candidates among the genes derived from the modified outlier analysis in lung cancer and breast cancer, respectively. Gene expression patterns were verified in cancer cell lines and patient tissues and the potential of these genes as therapeutic targets was identified by loss-of-function analysis and pharmacological treatment.
Materials and Methods
1. Modified Tukey’s fences outlier analysis
Expression data were downloaded from CCLE and TCGA to identify outlier genes in individual samples. We analyzed these data based on absolute expression (RNA-seq by expectation maximization [RSEM], Robust Multichip Average [RMA]) within the samples and outlier level (OL) was defined as the absolute expression divided by the interquartile range (IQR) of that gene. Each gene with a frequency of more than 1%, OL of more than 7.0, and modified Z-score of more than 10.0 was selected as a candidate outlier gene (S1 Fig.).
X is absolute expression in a sample; Q3, third quartile of the gene; Q1, first quartile of the gene; and IQR, Q3-Q1.
GraphPad Prism software (GraphPad, Inc., La Jolla, CA) was used to describe the outlier profiles for each sample.
2. nCounter gene expression assay
From the collected lung cancer formalin-fixed paraffin-embedded (FFPE) samples with sufficient tissue remaining, RNA was extracted using an RNeasy FFPE kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Samples processed using a customized nCounter gene expression panel, which allowed testing for 100 genes including outlier genes (S2 Table). Briefly, 200 ng of RNA was used, and the results were normalized using NanoString nSolver software. After performing image quality control using a predefined cutoff value, we excluded outlier samples using a normalization factor defined as the sum of positive control counts greater than threefold.
3. Cell lines
Breast cancer cell lines (BT-20, BT-549, HCC-38, MDA-MB-231, and ZR-75-1) and lung cancer cell lines (A549, HCC-15, HCC-44, HCC-1171, HCC-1833, NCI-H23, NCI-H1792, NCI-H1975, NCI-H2228, and SK-LU-1) were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. MCF7, Calu-3, and a normal lung cell line (BEAS-2B) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum at 37°C with 5% CO2. Breast cancer cell lines, lung cancer cell lines, and the normal lung cell line were obtained from the American Type Culture Collection or Korean Cell Line Bank. These cell lines were authenticated via short tandem repeat profiling before beginning a new series of experiments and were maintained in culture for < 3 months.
4. Quantitative reverse transcription–polymerase chain reaction
LRRK2 and TM4SF4 expression was examined using a PRISM 7900HT Fast Realtime PCR system (Applied Biosystems, Foster City, CA). The sequences of the primers used were as follows: LRRK2-F, 5′-CCTAAAGTGGAGAGTTTCAGTGC-3′; LRRK2-R, 5′-GATTGTCATAGAAGGAGGCAAGA-3′; TM4SF4-F, 5′-CTGGTGTTCTTGGGCCTGAA-3′; and TM4-SF4-R, 5′-GGTGAACATCGCAAATCGCT-3′. All reactions were performed in triplicate. RNA isolation and cDNA synthesis were performed using the RNeasy Mini Kit (Qiagen) and SuperScript III first-strand kit (Invitrogen, Carlsbad, CA) according to the manufacturers’ instructions.
5. Immunohistochemistry
Tissue sections (3 mm) were deparaffinized and rehydrated, and antigen retrieval was performed for 40 minutes in citrate buffer (pH 6.1) at 95°C. Diaminobenzidine was used as the chromogen, and the sections were counterstained with hematoxylin. The ScanScope AT automated slide processing system was utilized. The anti-TM4SF4 antibody (HPA046430, Sigma-Aldrich, St. Louis, MO) and anti-LRRK2 antibody (HPA014293, Sigma-Aldrich) were used for TM4SF4 and LRRK2 immunohistochemical staining.
6. Western blotting and fluorescence-activated cell sorting analysis
Western blotting was performed using antibodies against the following: LRRK2 (MJFF2; Abcam, Cambridge, UK), STAT3 (#4904, Cell Signaling Technology, Danvers, MA), phospho-STAT3 (Tyr705) (#9145, Cell Signaling Technology), AKT (#2920, Cell Signaling Technology), phospho-AKT (S473) (#4060, Cell Signaling Technology), cyclin D1 (#2978, Cell Signaling Technology), cyclin D3 (#2936, Cell Signaling Technology), CDK2 (#2546, Cell Signaling Technology), p27 Kip1 (#3686, Cell Signaling Technology), β-actin (sc-47778, Santa Cruz Biotechnology, Santa Cruz, CA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; sc-25778, Santa Cruz Biotechnology). For the fluorescence-activated cell sorting (FACS) analysis, anti-TM4SF4 antibody (MAB7998, R&D Systems, Minneapolis, MN) was used to stain cells.
7. Establishment of stable shRNA-expressing cell lines
A stable knockdown cell line, Calu-3, was established by infecting pLK0.1-based lentiviral particles with TM4SF4-specific target shRNAs (TRCN0000300821). Forty-eight hours after infection, the cells were divided into groups and exposed to 2 μg/mL puromycin for 14 days to eliminate uninfected cells.
8. Cell viability assay
Cells were seeded into 96-well plates at a density of 5.0×103 cells per well, allowed to adhere for 24 hours, and then treated with LRRK2-IN-1. After 48 hours, cell viability was measured using the WST-1 assay kit (EZ-3000, Daeillab Service, Seoul, Korea) according to the manufacturer’s instructions.
9. Statistical analysis
Statistical significances for clinicopathological characteristics were estimated using Fisher exact test and chi-squared test. An independent t test was used to analyze DNA copy number and DNA methylation status between outlier and others, quantitative reverse transcription–polymerase chain reaction (qRT-PCR), and cell viability. All tests were carried out using GraphPad Prism, with significance defined as p < 0.05.
Results
1. Discovery of cancer outlier genes
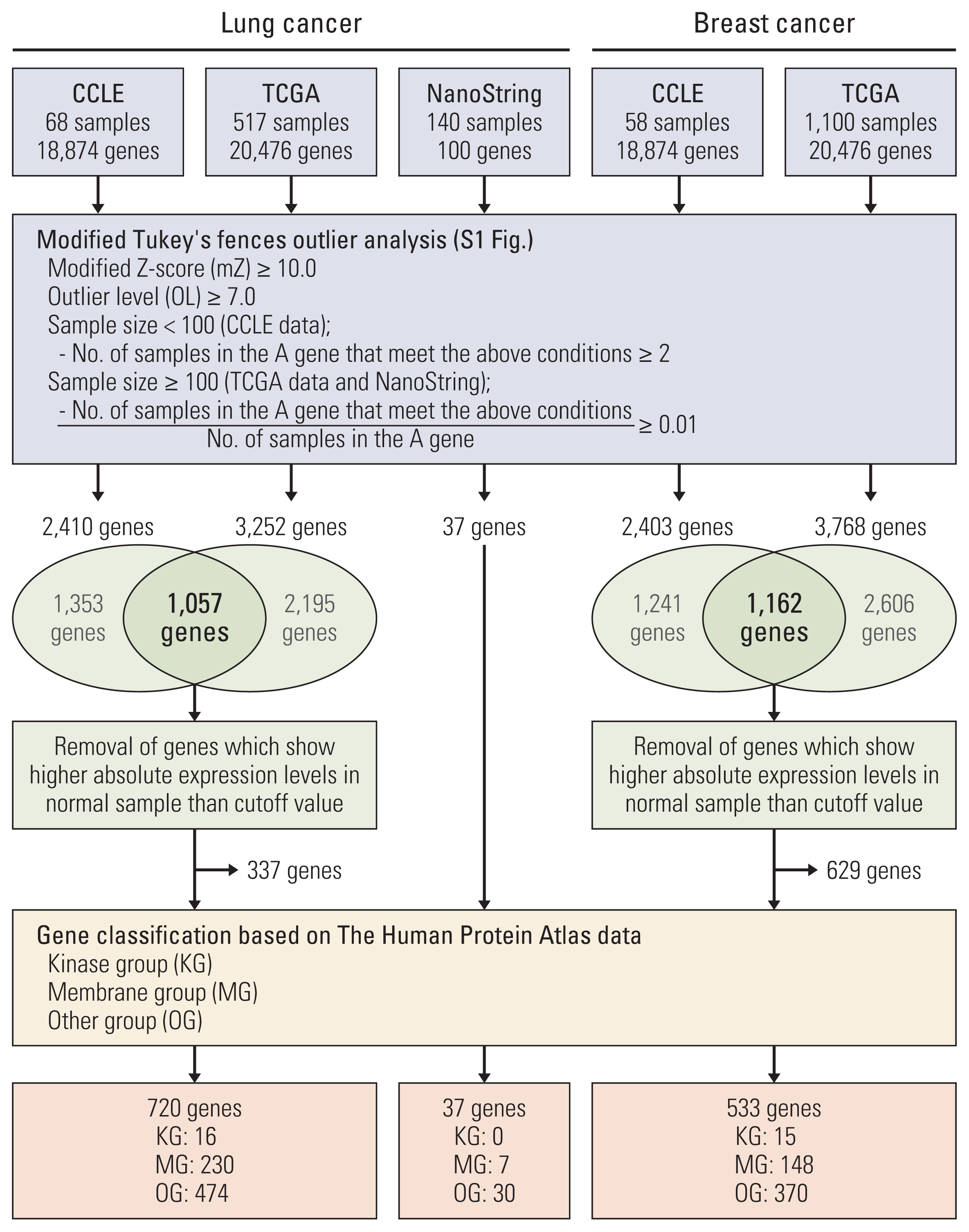
To discover genes with extreme outlier patterns in cancer, the CCLE and TCGA datasets were used for outlier analysis. We first sorted out genes that have samples with modified Z-score of more than 10.0. The modified Z-score of 3.5 or more is usually called an outlier. OL is defined as the number that the IQR is being multiplied to in the Tukey’s fences analysis. Usually, a OL of 1.5 or more is defined as an outlier [6]. Here, the genes were sorted with a cutoff at OL 7.0. And, in each dataset the gene with more than two outlier samples or 1 percent outlier samples was rescreened. Of the rescreened genes, we selected the genes found in both CCLE and TCGA. And, of the selected genes, the genes which show absolute expression in normal sample higher than cutoff in TCGA were removed to discover cancer-specific outlier genes. And analyzed genes were classified into groups of kinases and membranes in consideration of targeted therapy (Fig. 1, S1 Fig.).
To determine the outlier genes in lung cancer, we investigated the mRNA expression levels in 68 lung cancer cell lines from CCLE and 517 lung adenocarcinoma samples from TCGA. Total 2,410 and 3,252 genes were screened as outlier genes in CCLE and TCGA, respectively. Of these, 1,057 genes were common to CCLE and TCGA. In order to identify outlier genes as those whose absolute expression levels is high only in cancer samples, the genes were removed from outlier genes if their absolute expression levels in normal samples were higher than the cutoff value. Finally, 720 genes were selected as lung cancer outliers and classified into the kinase group, membrane group, and other group. Sixteen outliers were classified as the KG. Among the 16 kinase genes, three, CDK4, EPHA3, and RET, were classified as cancer-related genes based on THPA database. The breast cancer datasets from CCLE and TCGA were also analyzed and 533 genes were identified as breast cancer outliers. Of these, 15 genes belonged to the KG. Five of these kinase genes, CDK12, ERBB2, FGFR4, FLT3, and LRRK2, were classified as cancer-related genes based on THPA database (Table 1, Fig. 1, S3 Table).
2. The causative mechanism of the overexpression of outlier genes
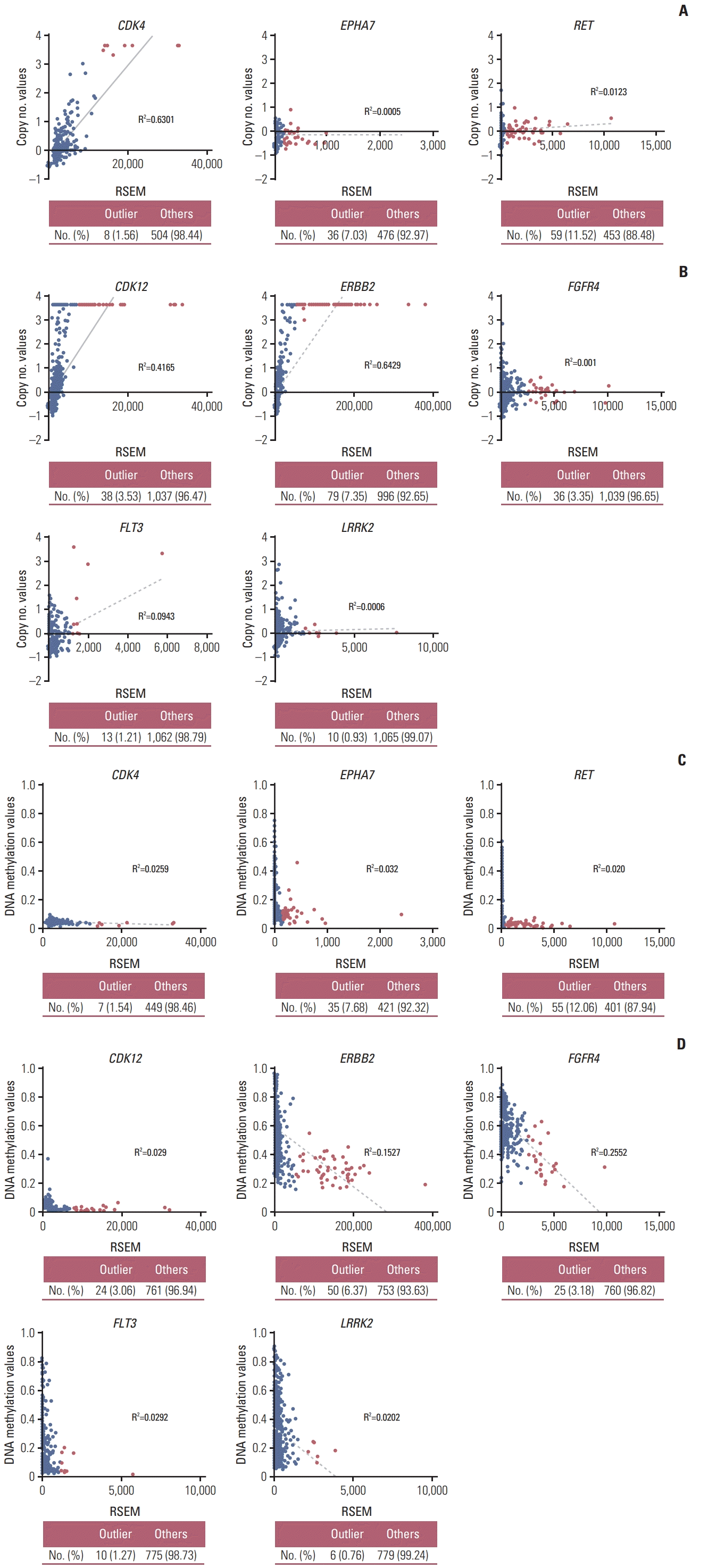
To investigate the causative mechanism of the overexpression of outlier genes, we compared the DNA copy number and DNA methylation status of each gene between outlier samples and others using TCGA datasets. In outlier samples, CDK4 in lung cancer, and CDK12, and ERBB2 in breast cancer showed a trend to have higher DNA copy numbers and DNA hypomethylation levels than in others. Their high overexpression seemed to be regulated by DNA copy number or DNA methylation status. And all samples with outlier FGFR4 and LRRK2 had lower copy number variation, whereas had DNA hypomethylation value than median value. Although the outliers FGFR4 and LRRK2 in breast cancer did not show DNA copy number alterations, these samples exhibited DNA hypomethylation, and the mechanism of their overexpression was predicted to be associated with their DNA methylation status (Table 1, Fig. 2, S4 Fig.).
3. Comparing outlier genes from modified Tukey’s fences method with outlier genes from COPA methods and differentially expressed genes (DEG) genes from DEGs analysis
To compare previous COPA with modified Tukey’s fences outlier analysis, we checked the overlapping genes that were screened as outlier gene using TCGA data. The number of kinase genes identified by the COPA method were 11 and 17 in lung cancer and breast cancer, respectively (S5A Fig., S6 Table). In the lung cancer dataset, two of the 11 kinase genes were classified as cancer-related genes in THPA database: CDK4 and EPHA8. Further, of the 17 kinase genes in the breast cancer dataset, CDK4, CDK12, ERBB2, and ROS1 were cancer-related genes. CDK4 in the lung cancer dataset as well as CDK12 and ERBB2 in the breast cancer dataset were selected as outlier genes by both the COPA method and present method. Notably, well-known oncogenes such as RET and FGFR4 were not sorted as outlier genes by the COPA method. However, RET in the lung cancer dataset as well as FGFR4 in the breast cancer dataset were selected as outlier genes by the modified Tukey’s fences method (S5B Fig.).
To check how the outlier genes are analyzed by traditional oncogene screening methods, we analyzed DEGs at TCGA dataset using edgeR R programming. DEG analysis is one of the widely used methods to identify novel biomarkers, such as oncogene, and it performs statistical analysis to screen genes with changes in gene expression between different groups. However, modified Tukey’s fences outlier analysis method screen genes that have outlier samples in a group. Among the outlier genes, 540 outlier genes of lung cancer and 436 outlier genes of breast cancer were suitable for DEGs analysis. Using a cutoff of 0.05 for p-value, 0.01 for false discovery rate, and |2.0| for fold change, we found that 357 (66.11%) and 225 (51.61%) genes were differentially upregulated in lung cancer outlier genes and breast cancer outlier genes, respectively. And although 183 of lung cancer outlier genes (33.89%) and 211 of breast cancer outlier genes (48.39%) were identified as downregulated genes or non-DEGs from DEGs analysis, we identified rare events with high expression, called outliers, in these genes. Known oncogenes, CDK4 and RET in lung cancer and ERBB2 and FGFR4 in breast cancer, were included in the upregulated genes [7–9]. CDK12 was also well-known oncogene, but CDK12 was found as the non-DEGs in DEGs analysis [10]. The DEGs are summarized in S3 Table. A volcano plot of the DEGs is presented in S7 Fig.
4. Function of TM4SF4 as an outlier gene in lung adenocarcinoma
To confirm the outlier gene expression, we conducted mRNA expression profiling of 140 lung adenocarcinoma samples using the NanoString nCounter system. These expression data were also analyzed by the modified Tukey’s Fences method. Among the total 100 genes including 50 outlier genes, 37 outlier genes were selected in the outlier analysis (Fig. 1, S8 Table). Seven genes were classified as MG. Of these, four genes, TM4SF4, MUC13, KCNH2, and RNF186, were sorted into outlier genes from the CCLE and TCGA data (S8 Table). Although TM4SF4 was sorted into upregulated gene in cancer compared to normal tissues by DEGs analysis, it was not attracting attention than other genes (S7 Fig.). However, TM4SF4 mRNA showed high absolute expression levels among outlier samples compared to other genes (S9A Fig.). In addition, TM4SF4 was classified into membrane group (S3 Table). TM4SF4 is a member of the tetraspanin family that is associated with cancer growth and motility [11], thus making it an attractive candidate as biomarker and target gene for targeted therapy if considering potential for antibody development. Based on the results of open data analysis and experimental data, we chose TM4SF4 for further studies.
To validate TM4SF4 as an outlier gene, we first confirmed the modified Z-score and OL in CCLE and TCGA data. The outlier cases were observed in 10 (14.71%) of the 68 lung adenocarcinoma cell lines in the CCLE data and in 52 (10.06%) of the 517 lung adenocarcinoma in TCGA data (S9B Fig.). And none of normal lung tissues was sorted as outlier cases (S10A Fig.). To determine the cause of TM4SF4 overexpression, we investigated the DNA copy number alterations and DNA methylation status in the TCGA data. TM4SF4 overexpression was found to be associated with DNA methylation status, but not with DNA copy number alteration (S11 Fig.). This association between the expression and DNA methylation status of TM4SF4 is consistent with previous observations in NSCLC cell lines [12].
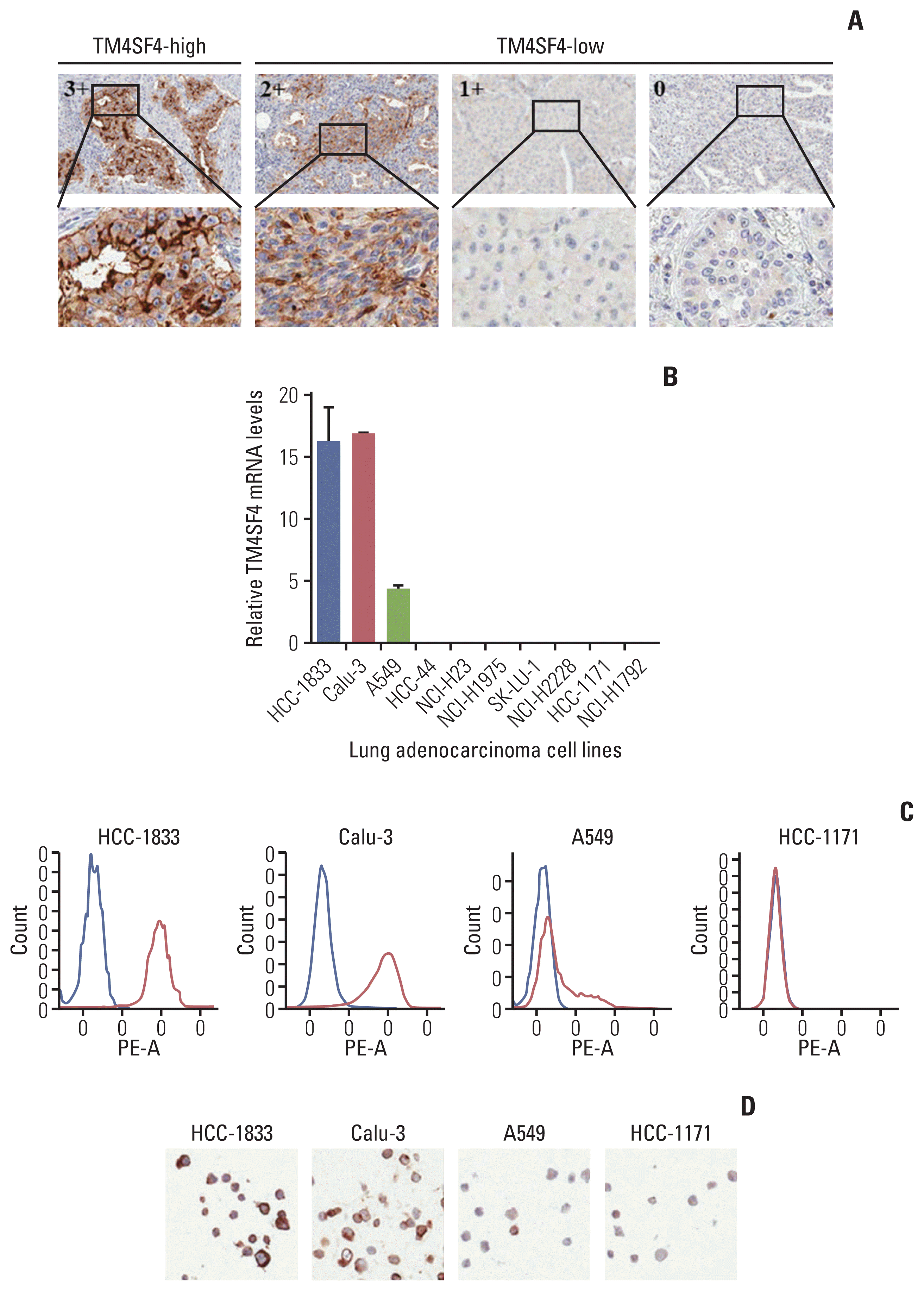
To confirm these informatics findings, we evaluated the mRNA and protein expression levels in lung adenocarcinoma tissues using a customized nCounter gene expression assay and tissue microarray (TMA). Eighteen of 140 adenocarcinomas (12.86%) showed outlier expression by nCounter gene expression assay (S9B Fig.). Among the 119 lung adenocarcinoma cases, five cases (4.20%) were scored for high TM4SF4 expression (3+) by TMA. TM4SF4 was predominantly expressed in the cell membrane, and the representative results of immunohistochemistry (IHC) staining are shown in Fig. 3A.
The clinical characteristics of these 119 patients are listed in S12 Table. Clinicopathological characteristics were not statistically significant between outliers and others. However, it may be difficult to assess the significance of clinicopathological characteristics according to TM4SF4 expression because the outlier sample sizes were small.
We also screened TM4SF4 expression in lung adenocarcinoma cancer cell lines by qRT-PCR, FACS, and IHC. TM4SF4 was overexpressed in HCC-1833, Calu-3, and A549 cells in the CCLE data (S9B Fig.). We also detected aberrant expression of TM4SF4 in HCC-1833, Calu-3, and A549 cells compared to that in other cells (Fig. 3B–D). Based on these results, further studies were carried out using A549 and Calu-3 cells.
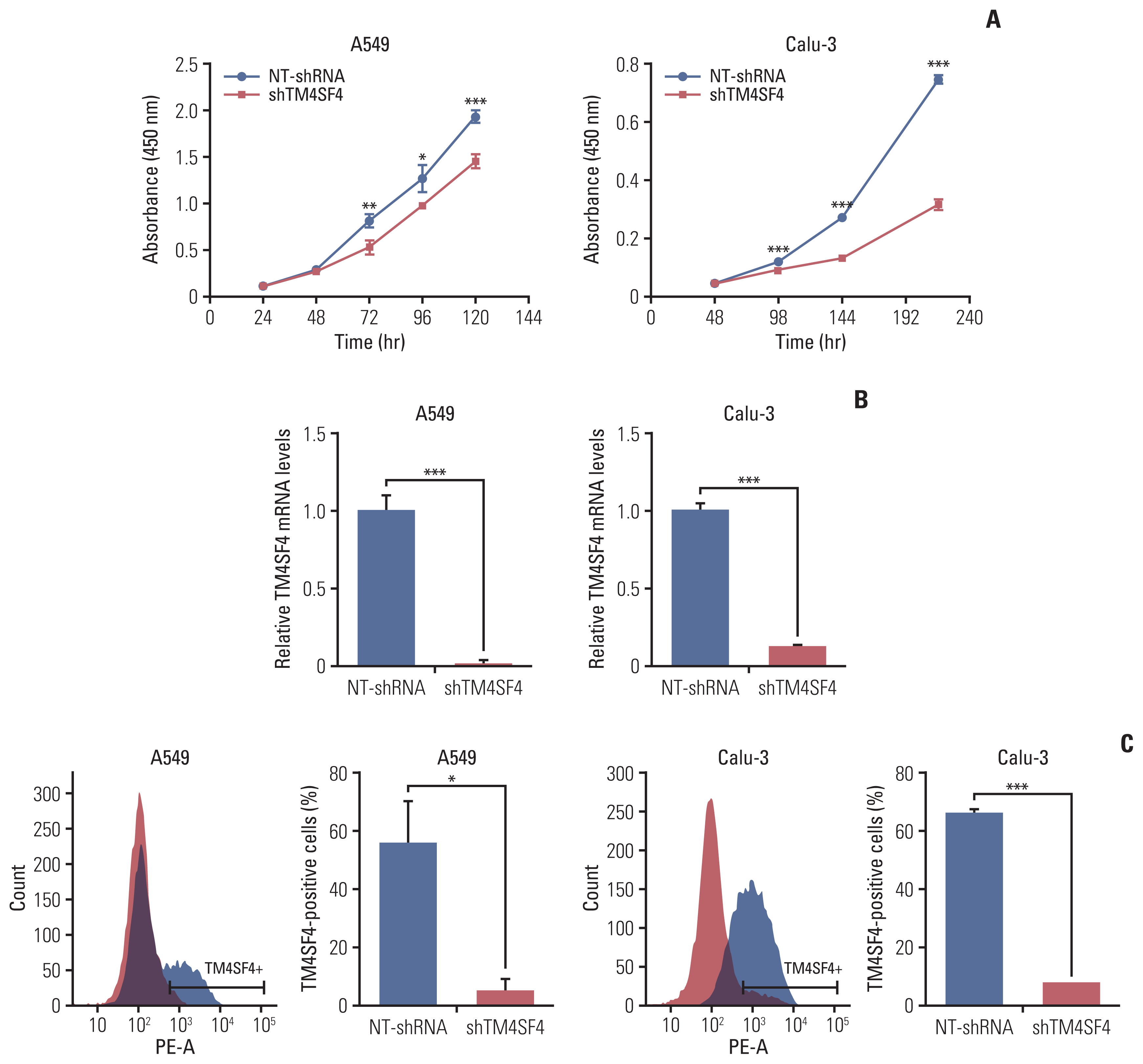
To investigate the role of TM4SF4 as an outlier gene in lung adenocarcinoma, we examined whether TM4SF4 regulates cell growth using an RNA interference (RNAi) system. A549 and Calu-3 cells were treated with siTM4SF4 or shTM4SF4, respectively. The results showed that cell growth in the si- or shRNA group was decreased compared to that in the control group. Knockdown efficiency was confirmed by qRT-PCR and FACS analysis. (Fig. 4, S13A–S13C Fig.).
We next conducted cell cycle analysis using propidium iodide staining to investigate the mechanism of action of TM4SF4 with respect to cell growth in A549 cells. A549 cells transfected with siTM4SF4 showed an increased G1 population and decreased G1/S checkpoint protein levels for cyclin D1, cyclin D3, and CDK2 (S13D–S13F Fig.). These results indicate that TM4SF4 knockdown induces cell cycle arrest in lung adenocarcinoma overexpressing TM4SF4.
5. Function of LRRK2 as an outlier gene in breast cancer
To find target biomarkers within breast cancer outlier genes, we first investigated potent and selective drugs to each gene in the kinase gene group. Seven genes had these potent and selective drugs (S14 Table). Of these genes, CDK12, ERBB2, FGFR4, FLT3 and RPS6KB1 are already well-known oncogenes in breast cancer, whereas LRRK2 have not been studied a lot in cancer [10,13,14]. LRRK2 activation by mutation is a well-known cause for Parkinson’s disease and many drugs are being studied and developed to inhibit LRRK2 activation [15,16]. For further study, we validated the target potential of LRRK2 considering drug repositioning.
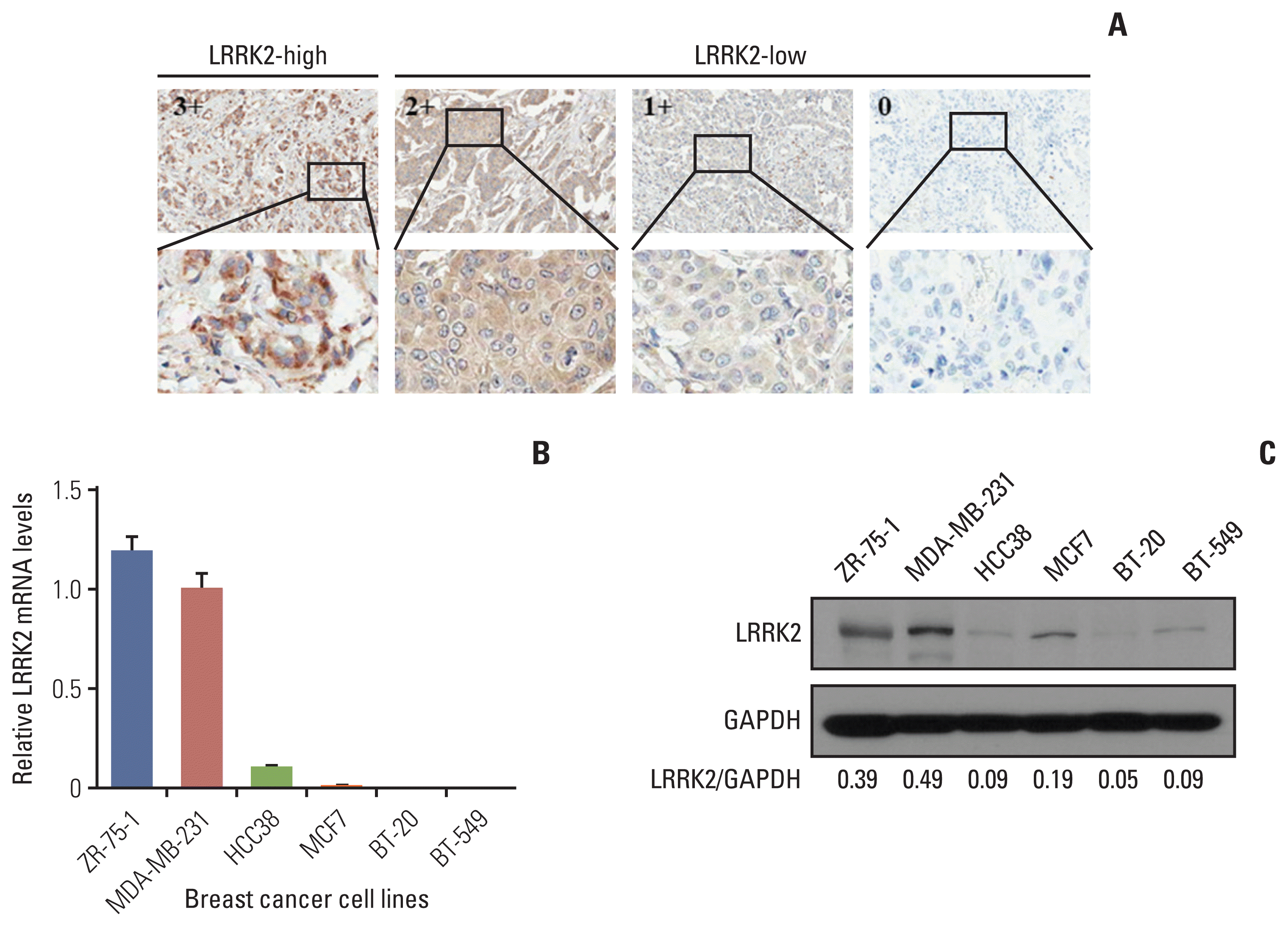
We first identified the modified Z-score and OL in CCLE and TCGA data. Outlier LRRK2 was observed in 4 (6.90%) of the 58 breast cancer cell lines and in 11 (1.00%) of the 1,100 breast invasive carcinoma (S9C Fig.). Normal tissue samples were not selected as outlier (S10B Fig.). We investigated the DNA copy number alterations and DNA methylation status in the TCGA data. The results showed that the DNA methylation status of LRRK2 showed a significant difference between outlier samples and others, but this was not observed for the DNA copy number value (Table 1, Fig. 2B).
To validate the analysis result using open data, we investigated the LRRK2 protein expression levels using TMA composed of human breast cancer tissues. Among the 417 cases analyzed, 10 cases (2.42%) were scored as high [3+] (Fig. 5A). We further investigated the clinical importance of LRRK2 in breast cancer. IHC scores for LRRK2 expression were as follows: outliers [3+] and others [0, 1+, and 2+]. Outlier samples were significantly correlated with age (p < 0.001) and estrogen receptor status (p=0.048). The differentiation, stages, and progesterone receptor status, and HER2 status showed no correlation (S15 Table). However, it may be difficult to assess the significance of clinicopathological characteristics because the outlier sample sizes were small.
For further functional studies, we analyzed the mRNA and protein expression levels in breast cancer cell lines by qRT-PCR and immunoblotting. The results indicate that the expression levels of LRRK2 in ZR-75-1 and MDA-MB-231 cell lines were higher than those in the other cell lines (Fig. 5B and C).
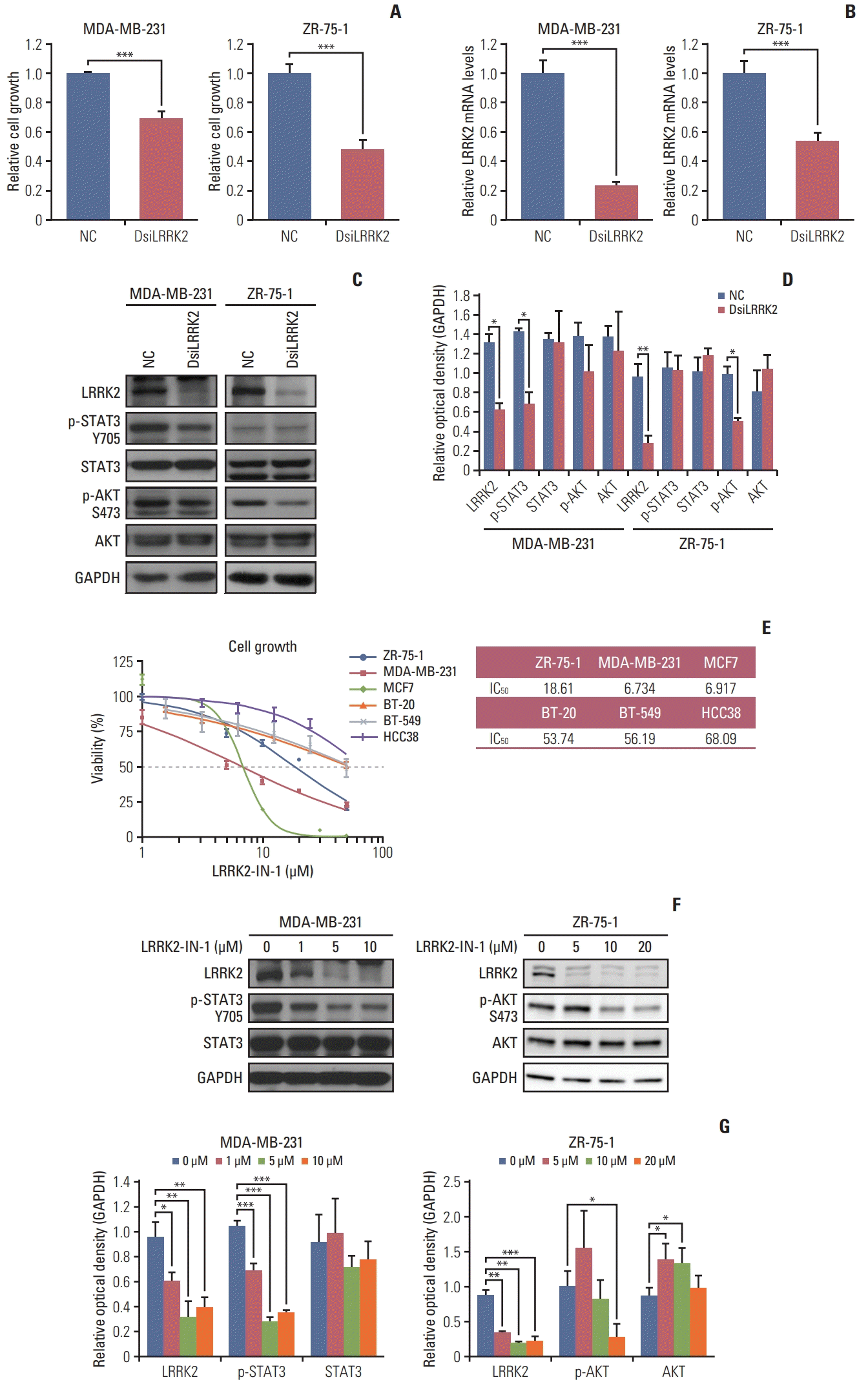
We examined whether LRRK2 regulates cell proliferation using an siRNA system. LRRK2-overexpressing MDA-MB-231 and ZR-75-1 cell lines were transfected with LRRK2 specific-target siRNA (DsiLRRK2) and negative control siRNA (NC). Treatment with DsiLRRK2 decreased the viability of both MDA-MB-231 and ZR-75-1 cells. Knockdown efficiency was confirmed by qRT-PCR and immunoblotting (Fig. 6A–D).
We further investigated LRRK2-mediated signaling related to cell proliferation. Treatment with DsiLRRK2 resulted in decreased levels of STAT3 phosphorylation (Tyr705) in MDA-MB-231 cells compared to those in the NC (Fig. 6C and D). Decrease in STAT3 phosphorylation following LRRK2 depletion has also been observed in type 1 papillary renal cell carcinoma cells SKRC39, with LRRK2 overexpression [17]. In contrast, treatment with DsiLRRK2 did not change the phosphorylation level of STAT3 (Tyr705) but decreased the phosphorylation level of AKT (S473) in ZR-75-1 cells (Fig. 6C and D) [18].
To evaluate the effect of LRRK2 pharmacological inhibition, we assessed the viability of breast cancer cell lines treated with different doses of LRRK2-IN-1, a potent and selective LRRK2 inhibitor [19]. Treatment with LRRK2-IN-1 dose-dependently reduced the viability of LRRK2-overexpressing cell lines, MDA-MB-231 and ZR-75-1, compared to that of LRRK2 under-expressing cell lines, BT-20 and BT-549 (Fig. 6E). Treatment with LRRK2-IN-1 resulted in decreased levels of phosphorylation of STAT3 (Tyr705) and AKT (S473) in MDA-MB-231 and ZR-75-1 cells, respectively (Fig. 6F and G). These results indicate that LRRK2 inhibition contributes to reducing cell proliferation and that the LRRK2 gene is a potential biomarker of pharmacologic responses in LRRK2-overexpressing breast cancer.
Discussion
Traditional analytical methods such as comparison of tumor and normal tissues have played a role in finding many biomarkers, but these methods have now reached a limit finding additional new ones [20]. A data point that deviates significantly from the group, called an outlier, may be an experimental error, but could also provide valuable information about abnormal characteristics. This study hypothesizes that an outlier reflects tumor heterogeneity and may represent a target gene for cancer therapy.
To discover cancer-specific outlier genes, we used modified Z-score and Tukey’s fences. These methods have the advantages of not having to follow a normal distribution and having less effect on extreme values, such as an outlier [6]. The modified Z-score is calculated from median and MAD, and these values were also used in COPA analysis. An outlier is typically defined as a Z-score value of 3.5 or more and as the value being multiplied to the interquartile range, calculated from Tukey’s fences analysis, of 1.5 or more [6]. But this study used 10.0 and 7.0 as cutoff values for screening extreme outlier genes. For selecting cancer-specific outlier genes, we removed the gene with samples that have higher absolute expression than cutoff in the normal dataset of the gene. Through this analysis, 720 genes and 533 genes were selected as outlier genes in lung and breast cancer, respectively. Of these genes, the candidate gene for targeted therapy or drug development is 16 kinase genes and 230 membrane proteins in lung cancer, and 15 kinase genes and 148 membrane proteins in breast cancer.
Various outlier analyses have been used to detect genes expressed at significantly higher levels in some samples than in others. This overexpression may occur through a variety of mechanisms such as genomic and epigenomic alterations [21]. Alteration in gene levels by these mechanisms may increase gene expression and activation, which is common in cancer cells. This can result in cancer cell growth or resistance to anticancer drugs [22]. Notably, ERBB2 in breast cancer and ALK in lung adenocarcinoma are representative genes overexpressed by genomic alteration such as DNA copy number alteration and chromosome rearrangement, respectively [23,24]. These genes play an oncogenic role by promoting tumor growth through multiple pathways such as those involving AKT, MAPK, and STAT kinase. Moreover, drugs targeting these genes have been successfully used in cancer therapeutic strategies [23,25]. Overexpression of claudin 18 isoform 2 in gastric cancer is associated with epigenetic derepression such as DNA hypomethylation of promoter. And, recently, anti-claudin 18.2 antibody is beginning to receive attention as new targeted therapy for advanced gastric cancer [26]. In our study, outlier-samples of cancer-related genes classified from THPA database, such as CDK4 in lung cancer and CDK12, ERBB2, and FLT3 in breast cancer, were associated with DNA amplification [7,10,13]. And outlier-samples of EPHA7 in lung cancer and FGFR4, and LRRK2 in breast cancer were associated with DNA hypomethylation. Expression of outlier-samples in RET in lung cancer was no associated with both DNA amplification and DNA hypomethylation. It seems that outliers overexpressed by chromosome rearrangement [8].
Through the modified Tukey’s fences outlier analysis, TM4SF4 was sorted as an outlier gene in both the TCGA and Nanostring validation datasets. TM4SF4 was identified as an upregulated gene form DEGs analysis although it was inconspicuous than other genes. And samples detected as outliers in TM4SF4 have extremely high expression compared to the other genes. TM4SF4 is a member of the transmembrane four superfamily, also known as the tetraspanin family. The members of this family are associated with cancer growth, invasion, and migration [11]. In addition, TM4SF4 is a suitable target gene to monoclonal antibody therapy because TM4SF4 protein is located in the cell membrane. We verified the TM4SF4 mRNA and protein expression in lung adenocarcinoma cell lines and lung adenocarcinoma patient tissues and found aberrant expression of TM4SF4 as an outlier gene in lung cancer. The cause of TM4SF4 overexpression in outlier samples seems to be associated with DNA methylation status but no DNA amplification [12]. Through a loss-of-function study using an RNAi system, we demonstrated that TM4SF4 knockdown led to reduction in cell growth. TM4SF4 seems to be involved in cell cycle checkpoints in A549 cells. Choi et al. [12] reported that TM4SF4 is overexpressed in radiation-resistant lung cancer cells, and it enhances activation of IGF1R signaling pathway and leads to increased tumorigenicity. Overexpression of TM4SF4 occurred dominantly in lung adenocarcinoma tissues compared with non-tumor tissues and large cell lung carcinoma [12]. TM4SF4 is also overexpressed significantly in hepatocellular carcinoma compared with TM4SF4 expression in non-tumor tissues, and TM4SF4 expression correlates with the tumor progression. Cell growth changes correlate with the expression of TM4SF4 [27]. TM4SF4 expression is related to tumor development in lung adenocarcinoma and hepatocellular carcinoma, so it can be used as a biomarker as well as a therapeutic target. However, additional functional studies on TM4SF4 are needed to understand the details of its role and to develop a TM4SF4 antibody as a therapeutic strategy.
To find target genes for targeted therapy in breast cancer, we investigated target drugs to each gene of kinase gene group. Of the fifteen kinase genes, seven genes had the potent and selective drugs. CDK12, ERBB2, FGFR4, and FLT3 are already well-known oncogenes with clinical significance in breast cancer. Interestingly, LRRK2 inhibitors have been developed for Parkinson’s disease therapy contrary to other drugs that were developed for cancer therapy [15]. LRRK2 is a member of the leucine-rich repeat kinase family and has diverse cellular functions and signaling pathways [28]. The exact role of LRRK2 is unknown, but a LRRK2 activating mutation, G2019S, has been implicated in the pathogenesis of Parkinson’s disease [16]. Whether the LRRK2-G2019S mutation increases cancer risk in patients with Parkinson’s disease is controversial [29,30]. However, LRRK2 activating mutations were not found in breast cancer (data not shown). Looyenga et al. [17] reported that chromosomal amplification of LRRK2 is related to the MET signaling pathway in sporadic type 1 papillary renal cell carcinoma. Amplification of LRRK2 was found in the TCGA breast cancer database, but it was not associated with mRNA expression. We found that LRRK2 was overexpressed in breast cancer using published data, breast cancer cell lines, and breast cancer tissues. LRRK2 overexpression in outlier samples seems to be associated with DNA hypomethylation. We next examined the effect of targeting LRRK2 using DsiLRRK2 and LRRK2-IN-1. Notably, inhibition of LRRK2 affected different pathways in MDA-MB-231 and ZR-75-1 cells. The LRRK2-mediated pathway is involved in the STAT3 pathway in MDA-MB-231 cells and AKT pathway in ZR-75-1 cells. Although LRRK2 influences different signaling pathways, LRRK2 knockdown showed the same results of reduced cell growth in both MDA-MB-231 and ZR-75-1 cells. Although LRRK2 was identified as non-DEG from DEGs analysis, we found that LRRK2 outlier samples in breast cancer have higher expression compared to normal tissues and present in approximately 1% of all breast cancers. These results suggest that LRRK2 is involved in controlling cancer cell growth through several pathways and may be useful as a biomarker in patients with breast cancer with LRRK2 overexpression.
In this study, we demonstrated the feasibility of using the modified Tukey’s fences outlier analysis method as a valuable preclinical platform for discovering personalized therapeutic targets. Additionally, the outlier analysis method suggested that TM4SF4 and LRRK2 are attractive targets for targeted therapy and that an anti-TM4SF4 antibody and small molecule inhibitors of LRRK2 can be used as targeted cancer drugs.
 XML Download
XML Download