

 PDF
PDF Citation
Citation Print
Print


서 론
면역글로불린 G4 연관질환(immunoglobulin G4 related disease, IgG4 연관질환)은 다양한 장기에 섬유화 염증을 일으켜 종양성 병변을 보이는 면역 매개 질환이다[1]. 특징적인 병리학적 소견을 보이는데 림프형질세포 침윤(lymphoplasmacytic infiltration), 소용돌이형 섬유화(storiform fibrosis), 폐색 정맥염(obliterative phlebitis)이 관찰되며, 면역염색에서 IgG4 양성형질세포가 증가되어 있다[1]. 흔히 침범되는 기관으로 췌장, 담관, 침샘, 눈물샘, 안와, 콩팥, 폐, 대동맥, 갑상샘 등이 있으며, 뇌와 척수의 경수막을 침범하는데 우리나라에서는 보고가 드물다[2-6]. 저자들은 IgG4 연관 두개강내 경수막염을 경험하여 보고한다.
Go to : 
증 례
50세 여자가 두통과 경련을 주소로 내원하였다. 경련은 좌측 팔의 간대성 움직임 뒤에 동반된 전신 강직간대경련이 2-3분간 있었다. 환자는 과거력에서 고혈압과 아스피린 유도 천식이 있어 기관지확장흡입제와 항고혈압제를 복용하고 있었다. 내원시 혈압은 133/70 mmHg, 맥박은 분당 87회, 호흡수는 분당 20회, 체온은 36.7oC였다. 다른 신체진찰에서는 특이소견이 없었다. 신경학적 진찰에서 의식은 명료하였고 뇌신경진찰에서 이상 소견은 발견되지 않았다. 좌측 손에서 medical research council 근력등급 4등급의 근력약화가 있었으나 수시간 뒤 호전되었고, 감각장애는 없었고, 심부건반사는 정상이었다. 그 외 다른 신경학적 이상 소견도 관찰되지 않았다. 내원 시 혈액 검사에서 헤모글로빈이 13.6 mg/dL였고 백혈구는 10,600/mm3 (호중구 5,600/mm3, 림프구 3,620/mm3, 단핵구 610/mm3, 호산구 450/mm3)로 증가되었다. 적혈구침강속도는 15 mm/hour, C-반응단백질은 0.718 mg/dL, 암모니아는 97 ug/dL이었다. 간기능검사와 신장기능검사는 정상이었다. B형간염바이러스항원, C형간염바이러스항체, 사람면역결핍바이러스항체, VDRL은 음성이었고 B형간염바이러스항체는 양성이었다. 항핵항체, 항중성구세포질항체, 항카디오리핀항체, 루푸스항응고인자는 음성이었고 류마티스인자, 안지오텐신전환효소는 정상범위였다. 면역글로불린(immunoglobulin; Ig) 검사에서 IgG는 1,106 mg/L, IgM은 73 mg/L, IgA가 79 mg/L로 정상 범위였고, IgG 서브클래스 검사에서 IgG1/IgG2/IgG3는 4,638.6/5235.9/295.6 mg/L로 정상범위였으나 IgG4는 3,000 mg/L 이상으로 크게 증가되어 있었다. 뇌척수액 검사에서 백혈구는 1/mm3, 단백 51.7 mg/dL, 포도당 수치는 36 mg/dL였으며, 세균, 결핵균, 진균은 배양되지 않았고, 악성세포도 관찰되지 않았다. 뇌자기공명영상에서 오른쪽 전두엽 영역의 덩이 모양의 결절성(nodular) 경막 비후가 주변 뇌부종과 함께 관찰되었으며, 왼쪽 두정엽과 전두엽 영역의 미만성(diffuse) 경막 비후가 관찰되었다(Fig. 1). 경수막 덩이의 감별진단을 위해 조직생검을 시행하였다. 병리 소견에서 급성 그리고 만성 염증과 섬유화가 일부 거대세포와 함께 관찰되었으며, IgG4 면역염색에서 다수의 면역글로불린 G4 양성 형질세포가 확인되었다(Fig. 2). 결핵종 감별 위해 시행한 항산균 염색에서 결핵균은 보이지 않았다. 치료를 위해 덱사메타손(5 mg/회, 4회/일)을 7일 동안 정맥주사하고 감량한 후, 경구 프레드니솔론(60 mg/일)으로 바꾸었으며 경과가 호전되어 퇴원하였다. 퇴원 후 1년 동안 프레드니솔론을 감량하면서 유지하였는데 경련의 재발은 없었고 좌측근력약화는 완전히 회복되었으며, 치료 기간 동안 다른 이상소견은 없었다. 1년 뒤 시행한 뇌자기공명영상에서 경수막 비후는 거의 호전되었다(Fig. 1).
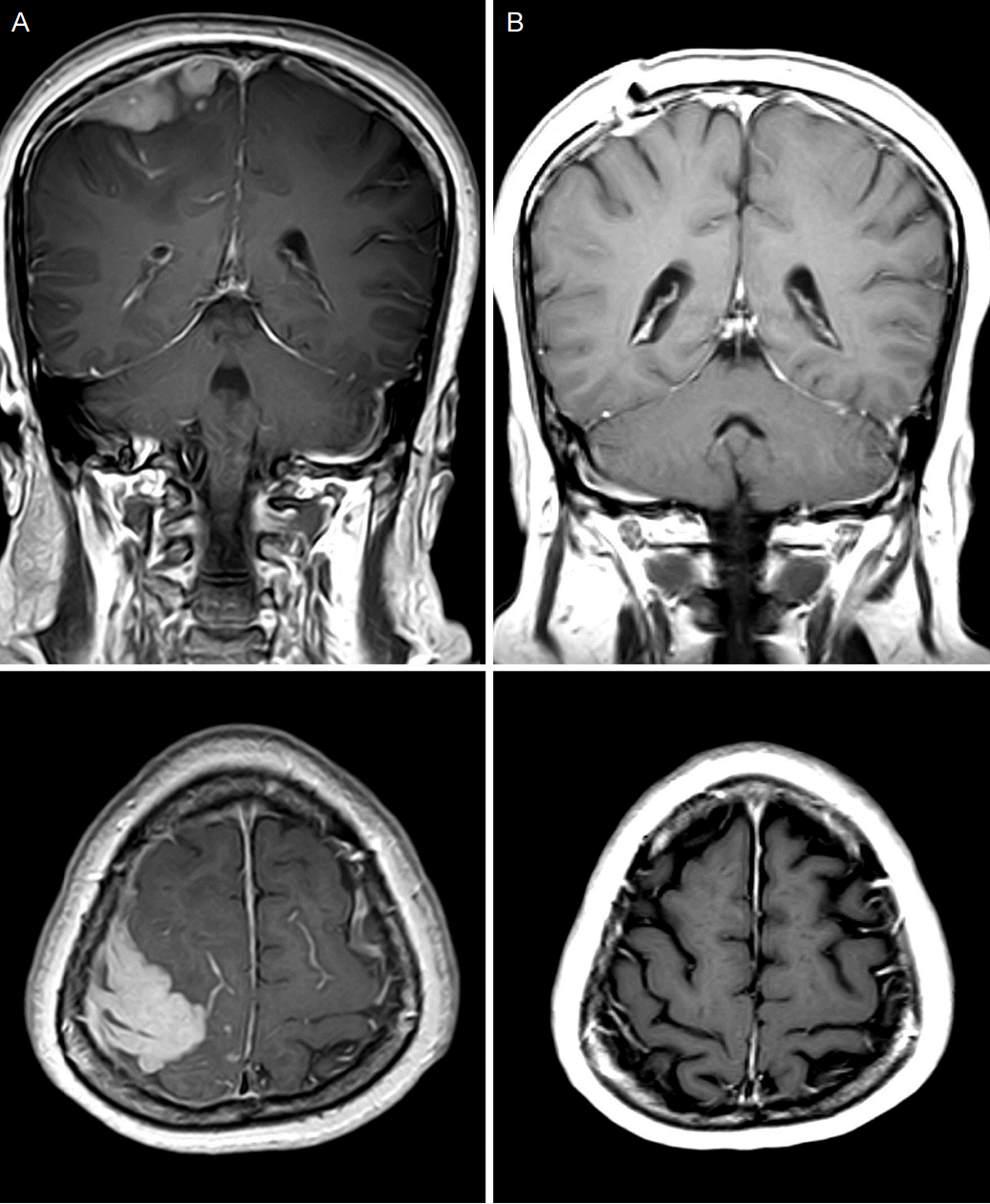 | Figure 1.Brain magnetic resonance imaging images, coronal view (upper row) and axial view (lower row). Initial T1-weighted images with gadolinium showed mass like nodular dural thickening with enhancement in the right frontal area and diffuse dural thickening in left frontal and parietal areas (A). After 1 year of treatment, T1-weighted images with gadolinium showed marked decrease of dural thickening and enhancement (B).
|
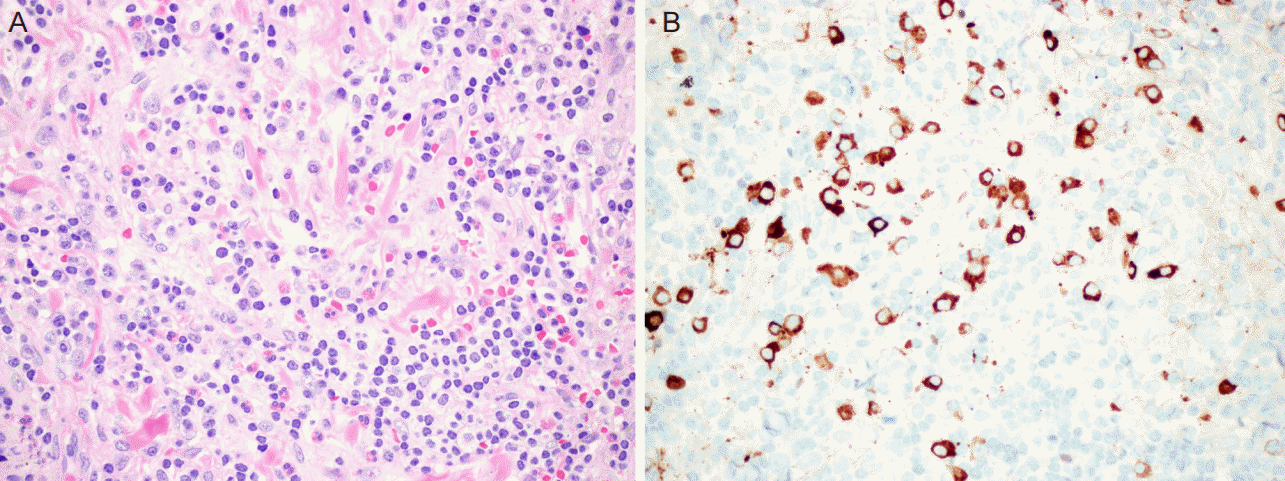 | Figure 2.Histpathology findings of meningeal mass biopsy. Hematoxylin and eosin staining showed intense lymphoplasmacytic inflammatory cell infiltrate with fibrosis (A, ×400). IgG4 immunohistochemistry showed numerous IgG4+ plasma cells (IgG4+/IgG+ plasma cell ratio > 40%) within the inflammatory infiltrate (B, ×400). IgG4, Immunoglobulin G4
|
Go to :
고 찰
IgG4 연관 질환은 2001년에 고농도 혈청 IgG4가 관찰되고 췌장의 면역조직화학검사에서 광범위한 IgG4 양성 형질세포가 관찰되는 경화성 췌장염 환자들을 보고하면서[7] 알려지기 시작한 질환이다. 2003년 췌장 이외에도 담관, 담낭, 위점막, 장점막, 침샘, 림프샘, 골수 등에도 IgG4 양성 형질세포가 발견되어 IgG4 연관 자가면역질환으로 명명되었다가 2012년 국제 다학제 연구 모임(Inter-National Multidisciplinary Study Group)에서 IgG4 연관 질환이라는 명칭이 제시되어 현재 사용되고 있다[8].
IgG4 연관 질환에서 2형 도움 T 세포(helper T cell type 2, Th2)로부터 나오는 interleukin (IL)-4, IL-5, IL-13이 증가되어 있고, IL-10과 transforming growth factors (TGF)-β를 생산하는 조절 T 세포가 관찰된다. IL-13과 TGF-β는 섬유화와 관련이 있으며, IL-4와 IL-10은 IgG4 클래스전환(class switch)에 관여하기 때문에 2형 도움 T 세포와 조절 T 세포가 중요한 역할을 할 것으로 보이지만 현재로서는 이를 증명하는 직접적 근거가 부족한 상태이다. 또한 혈액과 조직 내 IgG4가 증가되어 있으나 IgG4는 Fc (Fragment, crystallizable) 수용체와 보체 C1에 대해 친화력이 낮아 면역반응을 일으키는 능력은 다른 IgG 아형보다 적어 IgG4 연관 질환의 병인에 관한 연구는 더 필요하다[9].
IgG4 연관 질환의 임상진단 기준은 (1) 미만성/국소성 종창 또는 덩이에 의한 장기 침범, (2) 혈청 Ig G4 증가(>135 mg/dL), (3) 조직병리 소견에서 뚜렷한 림프구와 형질세포의 침윤과 섬유화가 관찰되고, IgG4 형질세포와 IgG 형질세포의 비가 40%를 초과하거나 현미경 고배율 시야에서 IgG4 형질세포수가 10개를 초과하여야 한다. 세 가지 조건을 모두 충족할 때 확진(definite), (1)과 (3)을 충족할 때 추정(probable), (1)과 (2)를 충족할 때 의심(possible)할 수 있다[10]. IgG4 연관 경수막염은 비후성 경수막염을 일으킬 수 있는 결핵, 매독 등에 의한 감염질환, 림프종, 전이뇌암 등의 종양질환, 사르코이드증, 베게너 육아종증을 포함한 항중성구세포질항체 연관질환등의 면역매개질환과의 감별이 필요한데, 본 증례에서는 조직검사를 통해 감염질환, 종양질환은 감별을 할 수 있었다. 또한 환자의 뇌자기공명영상에서 경막 비후 소견이 있었고. 항중성구세포질항체는 음성, 혈청 IgG4는 3,000 mg/L 이상으로 크게 증가되었고, 병리학적 소견에서 급성 그리고 만성 염증과 섬유화, 형질세포 침윤이 있으면서, 면역염색에서 IgG4 양성 형질세포가 확인되어 IgG4 연관 경수막염으로 진단할 수 있었다.
IgG4 연관 질환에서의 경수막 비후는 다양한 영상소견으로 나타난다. 두곳 이상의 비후가 함께 관찰되는 것이 단일 병변보다 많으며, 이 중 가장 흔한 소견으로 미만성 경막 비후가 39.5%, 두 번째로 결절성 경막 비후가 30.2%로 많다[6]. 본 증례에서는 오른쪽 전두엽의 조영증강되는 결절성 경막 비후와 왼쪽의 미만성 경막 비후가 함께 관찰되었다.
IgG4 연관 질환의 가장 중요한 치료제는 스테로이드이며 반응이 좋다[11]. 프레드니솔론(Prednisolone)을 40 mg/day 또는 0.6 mg/kg/day로 2-4주간 사용한 후 신체 검사, 혈액 검사, 방사선 검사 등을 종합하여 서서히 감량한다. 스테로이드의 감량 기간은 문헌마다 차이가 있으나 3-6개월간 감량하며[1,9], 길게는 3년까지 유지하자는 주장도 있다[12]. 본 증례에서는 스테로이드를 1년간 지속 사용하였고, 추적 관찰 기간 동안 재발이나 다른 장기의 침범은 없었다. 그러나 IgG4 연관 질환 환자의 약 30%에서 스테로이드 투여 후 감량하는 도중에 혹은 감량한 뒤 질환이 재발되거나 스테로이드 부작용으로 치료에 실패할 수 있어 azathioprine [5], mycophenolate mofetil [13], cyclophosphamide [5], methotrexate [3]등의 다른 면역억제제를 사용할 수 있다. 스테로이드나 다른 면역억제제에도 효과가 없어, B 세포 활성 억제와 관련하여 리툭시맙(rituximab)을 사용한 뒤 호전된 경우도 있다[14]. 본 증례의 경우에는 다른 치료제의 사용 없이 스테로이드를 사용하였으며, 1년 후 시행한 MRI상에서 경수막 비후의 호전이 관찰되었다.
IgG4 연관 경수막염은 경수막의 비후로 인한 덩이 효과로 신경학적 증상을 일으키고 스테로이드 사용시 반응이 좋은 질환으로, 비후성 경수막염이 있을 때 반드시 감별진단에 포함되어야 한다.
Go to :
 XML Download
XML Download