

 PDF
PDF Citation
Citation Print
Print


Introduction
Breast cancer (BC) remains the cancer type with the highest morbidity among women, with 1.67 million diagnoses and 521,900 deaths in 2012 alone [1]. In recent years the average 5-year survival of BC patients has risen significantly owing to advances in adjuvant and local therapeutic treatments, but in individuals with metastatic disease, such survival rates remain poor even following radical surgical treatment [2]. BC is a highly heterogeneous disease, with many recent efforts having been made to group BC patients according to a number of phenotypic parameters including hormone receptor (HR) expression status and human epidermal growth factor receptor 2 (HER2) expression levels, leading to the definition of BC subtypes including luminal A, luminal B, HER2, and triple-negative BC [3]. Between 60% and 75% of BC cases are found to be HR-positive upon diagnosis [4]. The prognosis of BC patients is influenced by a range of different parameters, with HR-positive BC being known to have a significantly better average prognosis relative to HR-negative BC [5,6]. BC tumors that are positive for estrogen receptor (ER) are dependent upon active estrogen levels for growth, and as such patients can be treated with estrogen blockers such as tamoxifen or with inhibitors of estrogen production, leading to a better patient prognosis. Similarly, patients with HER2-positive BC often respond well to the monoclonal antibody trastuzumab, leading to significantly improved survival outcomes [7]. However, trastuzumab resistance also frequently manifests among these patients and is associated with significantly poorer survival outcomes. Triple-negative BC, in contrast, lacks any available targeted therapeutics and is thus associated with a poorer prognosis than other disease subtypes [8]. These findings emphasize the importance of identifying prognostic biomarkers and therapeutic targets which may allow for the better management and treatment of HR-negative BC patients.
Atypical protein kinases of the right open reading frame (RIO) family are present in almost all forms of life [9,10], with family members including RIOK1, RIOK2, and RIOK3. Both RIOK1 and RIOK2 are non-ribosomal proteins that are nonetheless essential for regulating ribosomal RNA biogenesis and cell cycle progression, with the depletion of either of these proteins in yeast having been shown to result in impaired 20S pre-ribosomal RNA processing. Similarly, RIOK2 is essential for 18S pre-rRNA production in human cells, whereas 21S pre-rRNA processing requires the activity of RIOK3 [11]. Proteins that regulate ribosomal biogenesis are known to have a profound impact of progression through the cell cycle, with RIOK1 depletion having been shown to result in S phase and mitotic arrest [12]. RIO kinases are also known to be important in the regulation of a wide range of disease types [13,14], with their overexpression having been previously detected in both non-small cell lung cancer (NSCLC) and colorectal cancer (CRC) [15]. In these contexts, RIOK1 has been shown to influence the proliferative, migratory, and invasive activity of tumor cells, although its exact mechanistic role in this context has not been fully [16]. Whether RIOK1 is similarly overexpressed and/or functionally important in BC remains uncertain, and further research is thus warranted to explore its relevance in this disease context.
Given that the biological function of RIOK1 in BC remains unclear, in the present study, we first evaluated the expression of RIOK1 in BC and then investigated the correlation between the RIOK1 expression and patient clinicopathological characteristics. We also evaluated the biological roles of RIOK1 by modulating its expression in BC cell lines. We found that knocking down RIOK1 was able to inhibit invasion, proliferation, G2/M cell cycle progression, and angiogenesis, while promoting cellular apoptosis via the phosphoinositide 3-kinase (PI3K)/AKT and mitogen-activated protein kinase (MAPK)/ERK pathway. Overexpression of RIOK1, in contrast, yielded the opposite phenotype. Bioinformatics analyses revealed that miR-204-5p was able to regulate the expression of RIOK1. Collectively, we found that RIOK1 plays an oncogenic role in BC and may represent a potential treatment target for BC patients.
Materials and Methods
1. Patient sample collection
Sixty BC patient tissue samples were collected from patients undergoing surgical resection for quantitative real-time polymerase chain reaction (qRT-PCR) and western blot in the general surgery department of the Affiliated Hospital of Nantong University between 2018 and 2019. Following collection, tissues were snap-frozen prior to use. In addition, 166 paraffin-embedded BC patient tissue blocks were collected in the department of pathology between 2010 and 2013 and were used for the retrospective study. All BC patient tumor samples had been independently evaluated by two pathologists, with differentiation, p53 expression levels, and HR/HER2 status being determined in light of the World Health Organization classification criteria. Patients that had undergone pre-surgical chemotherapy or radiotherapy were excluded from this analysis.
2. Immunohistochemistry and scoring
The expression of RIOK1 was assessed immunohistochemically in the 166 paraffin-embedded tumor tissue samples (5-µm-thick sections) discussed above using a tissue microarray approach, with each tissue sample having a core diameter of 2 mm. Sections were first treated with dimethyl benzene to achieve deparaffinization, after which an ethanol gradient was used to dehydrate samples. Antigen retrieval was conducted by heating samples at 100°C for 4 minutes and then at 95°C for 10 minutes in sodium citrate buffer (10 mM sodium-citrate mono-hydrate, pH 6.0), after which samples were allowed to cool to room temperature over 20 minutes prior to being washed with phosphate buffered saline (PBS). A 0.3% H2O2 solution was then applied to all samples for an additional 20 minutes in order to inhibit endogenous peroxidase activity, after which samples were probed overnight using anti-RIOK1 (1:100, Immunoway Group, Plano, TX) at 4°C. A two-step reagent kit (horseradish peroxidase [HRP] anti-mouse/rabbit IgG, Dako, Santa Clara, CA) was then used to detect this primary antibody, with diaminobenzidine (Dako) and hematoxylin counterstaining (Dako) being used to evaluate RIOK1 expression levels in these tissue samples. Two pathologists that had been blinded to patient outcomes next independently assessed RIOK1 staining in each tissue samples, scoring tissues according to staining intensity (with scores of 0, 1, 2, and 3 corresponding to no, weak, moderate, and strong staining, respectively) and the percentage of RIOK1-positive cells (with scores of 0, 1, 2, and 3 corresponding to 0%-30%, 31%-60%, 61%-80%, and 81%-100% RIOK1-positive, respectively). The product of these scores was then used to assess RIOK1 staining intensity, with an overall score of 0-3 being considered “low” and a score of 3 or higher being considered “high.”
3. qRT-PCR
TRIzol (Thermo Fisher Scientific, Waltham, MA) was used to isolate total RNA from 60 BC patient tissue samples after which a spectrophotometer (NanoPhotometer, IMPLEN, Munich, Germany) was used to quantify the RNA contents in individual samples. Next, the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) was used to prepare cDNA from 10 µL of each RNA sample using the following thermocycler settings: 42°C for 60 minutes and 70°C for 5 minutes. A Roche LightCycler 480 (Roche, Basel, Switzerland) was used to conduct qRT-PCR analyses of three replicate 2 µL cDNA samples, with individual reactions also containing 10 µL SYBR Green I Mix (Roche), 0.5 µL each of forward and reverse primers, and 7 µL nuclease-free H2O. Thermocycler settings were as follows: 95°C for 5 minutes; 45 cycles of 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. Primers used in this study were as follows: RIOK1 (F, 5′-CCTTGGATTCTGATAACTGGAC-3′; R, 5′-AGGAAAATGGTGAAAACTTGG-3′), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; F, 5′-CGCTGAGTACGTCGTGGAGTC-3′; R, 5′-GCTGATGATCTTGAGGCTGTTGTC-3′). GAPDH expression was used for normalization purposes, with the 2−ΔΔCT approach used to assess relative RIOK1 expression levels in samples.
4. Western blot
RIPA lysis buffer containing protease inhibitors was used to lyse and BC tumor tissue samples, which were then spun for 20 minutes at 12,000 rpm at 4°C. Supernatants were then collected, with a BCA kit (Beyotime Institute of Biotechnology, Nantong, China) being used to quantify the protein contents within each sample. Samples were then boiled for 15 minutes in sodium dodecyl sulfate sample buffer, after which equal quantities of protein were separated via sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were in turn blocked for 2 hours at room temperature with 5% skim milk in TBST. Blots were then probed with rabbit polyclonal anti-RIOK1 (1:1,000, Proteintech, Wuhan, China), anti–E-cadherin (1:1,000, Proteintech), anti-vimentin (1:1,000, CST, Danvers, MA); anti–N-cadherin (1:1,000, CST); PI3K (1:1,000, CST); p-AKT (1:2,000, CST); AKT (1:1,000, CST); cyclin B1 (1:500, Proteintech); p-ERK1/2 (1:1,000, Abcam, Cambridge, UK); ERK1/2 (1:2,000, Abcam). After washing in TBST, the membran washed thrice in TBST and incubated for 2 hours with appropriate HRP-conjugated secondary antibodies (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA). Enhanced che-miluminescence (Thermo Scientific) was then employed for protein detection, with analyses being repeated in triplicate.
5. Cell culture and transfection
BC cell lines MDA-MB-231 (HR-negative) and MCF-7 (HR-positive) were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). MDA-MB-231 were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (GIBCO-BRL, Invitrogen), 100 µg/mL penicillin and 100 U/mL streptomycin (Shanghai Genebase Gen-Tech, Shanghai, China). MCF-7 cells were maintained in RPMI (Invitrogen) supplemented with 10% fetal bovine serum (GIBCO-BRL, Invitrogen), 100 ug/mL penicillin, and 100 U/mL streptomycin. All cells were within a humidified atmosphere containing 5% CO2 at 37°C. MDA-MB-231 cells and MCF-7 cells were transfected with Lipofectamine 3000 Reagent (Invitrogen) following the manufacturer’s protocol. Transfection efficiency was evaluated by western blot and quantitative polymerase chain reaction.
6. Flow cytometry
Flow cytometry analysis was performed detecting cell cycle distribution and cell apoptosis according to the manufacturer’s protocol. Briefly, cells were trypsinized and fixed in centrifuge tubes containing 0.5 mL of 70% ethanol for at least 6 hours at 4°C, and then the suspension was centrifuged for 5 minutes at 1,000 rpm. Cell pellets were resuspended in 5 mL of PBS for approximately 30 seconds and centrifuged at 300 ×g for 5 minutes, then resuspended in 1 mL of propidium iodide staining solution and kept in the dark at 37°C for 10 minutes. Samples were analyzed by a flow cytometer (BD, Franklin Lakes, NJ). The percentage of the cells in G0-G1, S, and G2-M phase were collected and counted. When analyzing cell apoptosis, cells were washed by PBS and resuspended at a concentration of 1×106 cells/mL. Then an Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, Oxford, UK) was used following the manufacturer’s protocol. After incubation in the dark at room temperature for 20 minutes, the samples were immediately analyzed by a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Each assay was performed in triplicate.
7. Cell migration, invasion assay, and wound healing assay
For cell migration and invasion assays, the cells were performed using a transwell system that incorporated a polycarbonate filter membrane. The transfected cells (5×104 cells per well) were plated in the upper chamber containing 200 µL of serum-free media. The lower chambers contained 10% fetal bovine serum. After 24 hours of incubation, chambers were selected and fixed with paraformaldehyde, then stained with crystal violet for migration assays. For invasion assays, the filters were precoated with Matrigel (BD) and DMEM or RPMI media mixture in a ratio of 1:6. We selected chambers of MDA-MB-231 after 24 hours and those of MCF7 after 48 hours. The number of migrating or invading cells was counted on the captured images.
For wound healing assay, cells were plated and transfected on 6-well plates. After cells reached confluence, the cells were incubated in serum-free DMEM or RPMI media. Then an artificial scratch of the cells was wounded with a 10 µL pipette tip. Then the cells were washed by PBS twice and serum-free medium was added for a further 24 hours. Each experiment was repeated three times.
8. Cell proliferation assays
We used Cell Counting Kit-8 (CCK-8; Dojindo, Tokyo, Japan), colony formation, and EdU assays to estimate cell ability of proliferation. Three thousand transfected cells were seeded in the 96-well plates and added 10 µL solution per well; the absorbance was read on 450 nm for 24 hours, 48 hours, 72 hours, and 96 hours. For the colony formation assay, a total number of 800 transfected BC cells were plated in 6-well plates and cultured for about 2 weeks. Cell colonies were fixed with 4% methanol and stained with crystal violet. Colonies were counted and each experiment was repeated 3 times. EdU assay (5-ethynyl-20-deoxyuridine) was performed with a commercial kit (Ribobio, Guangzhou, China). The red staining represented proliferating cells and blue staining represented cell nucleus.
9. Tube formation assay
Two hundred microliters of Matrigel (BD) was plated in 24-well plates and incubated at 37°C for 30 minutes. When it became gel, 8×104 HUVECs were added in plates in two groups: supernatant from siNC transfection or siRIOK1 transfection of MDA-MB-231. After incubation at 37°C for 6 hours, tube formation was observed by microscope and saved.
10. Luciferase reporter assay
The wild type or mutation sequences within the predicted 3′ untranslated region binding sites of the RIOK1 were transfected into 293T cells along with miR-204 mimics or NC, carrying a luciferase reporter plasmid. After 48 hours, the luciferase activity was measured by Dual-Luciferase Reporter Assay Kit (Promega, Madison, WI).
11. Tumor xenografts
Four-week-old female nude mice were injected with MDA-MB-231 cells (1×107) with siRIOK1 or NC subcutaneously. After first injection, we transfected them in vivo each 5 days. The volume of xenograft tumors was measured every 5 days. After 30 days, the mice were executed and tumors were taken out for weighing.
12. Bioinformatics analysis
RIOK1 expression levels in a large BC patient cohort were assessed using the Oncomine database (https://www.oncomine.org) using the search terms “RIOK1,” “Cancer VS. Normal/Cancer Analysis,” and “Breast Cancer.” Data were compared based upon log2 median-centered intensity in Oncomine microarray datasets. Kaplan-Meier Plotter (http://kmplot.com/analysis/) was further used to assess the relationship between RIOK1 expression levels and BC patient survival outcomes, yielding a survival curve for 3,951 BC patients and providing the corresponding p-value describing the relationship between RIOK1 expression and BC patient prognosis.
13. Statistical analysis
All statistical testing was conducted using SPSS ver. 20.0 (IBM Corp., Armonk, NY), and GraphPad Prism 6.0 (GraphPad Software Inc., San Diego, CA) was used to generate figures. Data are mean±standard deviation and were compared via Student’s t tests, Kaplan-Meier survival analyses, chi-square tests, and Cox regression analyses as appropriate. For univariate and multivariate analyses of factors associated with BC patient prognosis, Cox proportional hazards regression models were used, with the results of these analyses reported as hazard ratios. p < 0.05 was the significance threshold.
14. Ethical statement
The xenograft mice were performed with the Afliated Hospital of Nantong University Animal Ethics Committee and according to the institutional guidelines.
The ethics committee of Nantong University Affiliated Hospital approved the present study, with all patients having provided written informed consent to participate.
Results
1. RIOK1 expression of BC patients in database
We began by analyzing the Oncomine database to assess RIOK1 expression levels in BC patients. This analysis revealed that patients with ER- or progesterone receptor (PR)–negative BC had higher average RIOK1 expression levels than did patients who were ER- or PR-positive (p < 0.001) (Fig. 1A). The similar analysis showed the expression of RIOK1 was different depended on the subclass of BC (Fig. 1B). Furthermore, we found using Kaplan-Meier Plotter that BC patients with higher RIOK1 expression had a poorer prognosis than did patients with lower expression of this gene (p < 0.001) (Fig. 1C).
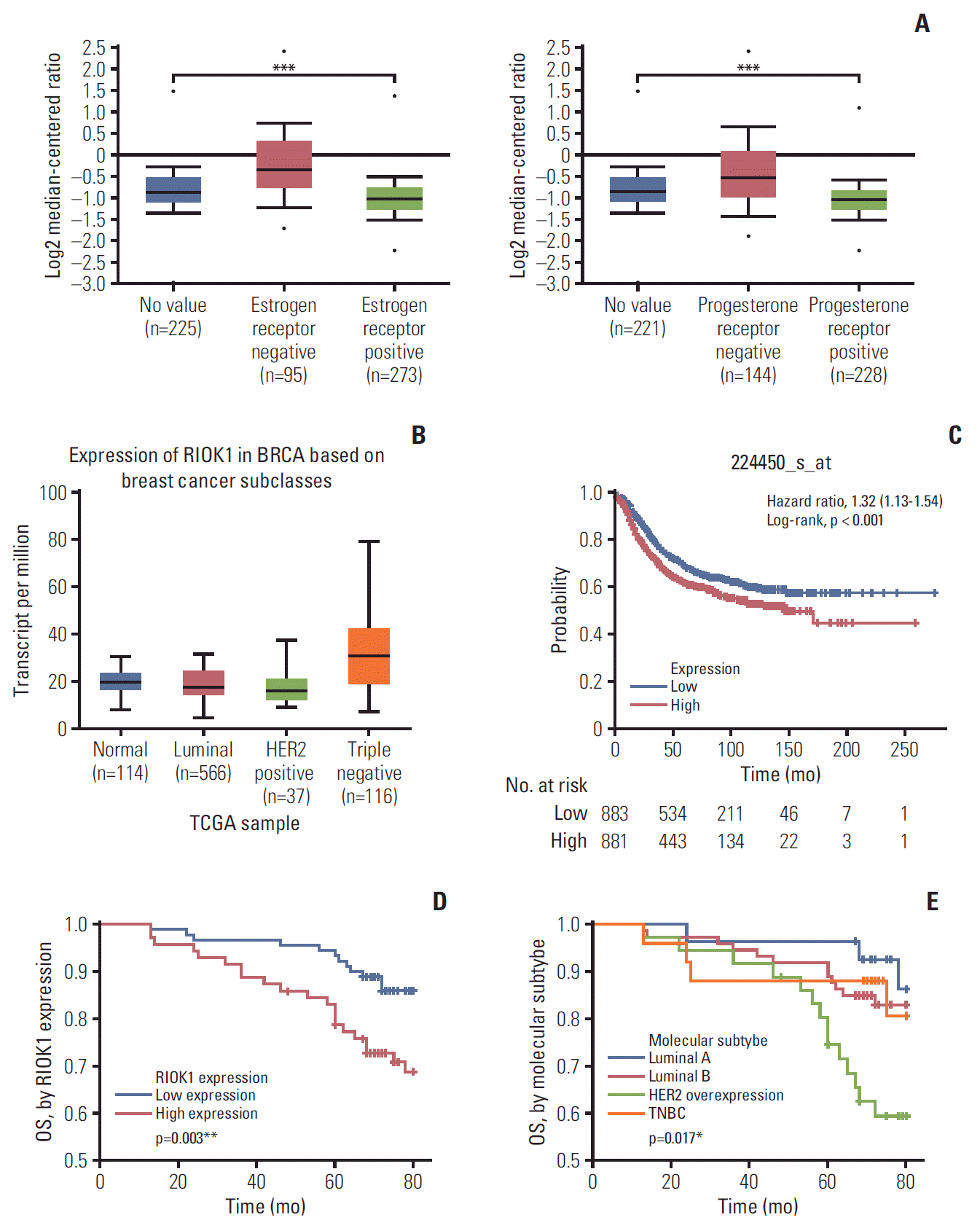
Fig. 1.
The expression of RIOK1 was increased in breast cancer (BC) tissues and associated with poorer patient prognosis. (A, B) Expression of RIOK1 level in patients with hormone receptor (HR) status and different BC subclasses in The Cancer Genome Atlas (TCGA) database. (C) Kaplan-Meier survival curve of patients with BC from the TCGA database. (D) The overall survival (OS) of RIOK1-high patients (red) was significantly lower than that of RIOK1-low patients (blue). (E) The OS of HR-negative patients (green/orange) was significantly lower than that of HR-positive patients (red /blue). *p < 0.05, **p < 0.01, ***p < 0.001.
![]()
2. RIOK1 is overexpressed in HR-negative BC tissues and correlates with clinicopathological parameters
We first quantified RIOK1 expression via western blot in five HR-positive and five HR-negative BC tumor samples and immunohistochemistry staining. This analysis showed that the expression of RIOK1 at the protein level was significantly higher in HR-negative BC tissue samples (Fig. 2A, B, and D). We further confirmed that RIOK1 expression was higher at the mRNA level in HR-negative BC tissue samples by comparing 40 HR-positive and 20 HR-negative samples via qRT-PCR (p < 0.001) (Fig. 2C). To explore the relationship between RIOK1 expression levels and BC disease progression using a tissue microarray of 166 BC patient samples. Of these 166 samples, 91 (54.8%) exhibited high RIOK1 immunohistochemical expression, while the remaining 75 (45.2%) are RIOK1-low (Table 1). Tumors that of a higher grade had significantly increased RIOK1 protein expression levels (p=0.010) (Fig. 2E). In these samples, we found that RIOK1 expression was higher in HER2+ and triple-negative BC patient tumor samples relative to those from patients with luminal A or B disease based on HR status (p < 0.05). No relationship was observed between RIOK1 expression and patient age, tumor diameter, lymph node metastasis, or Ki67 expression. p53 staining results from the pathology department also revealed a significant association between RIOK1 and P53 levels in these BC patient tissue samples (p=0.008). Database also suggested a potential correlation between RIOK1 and p53 (Fig. 2F). Moreover, elevated RIOK1 expression was associated with an 80-month survival duration (p=0.011).
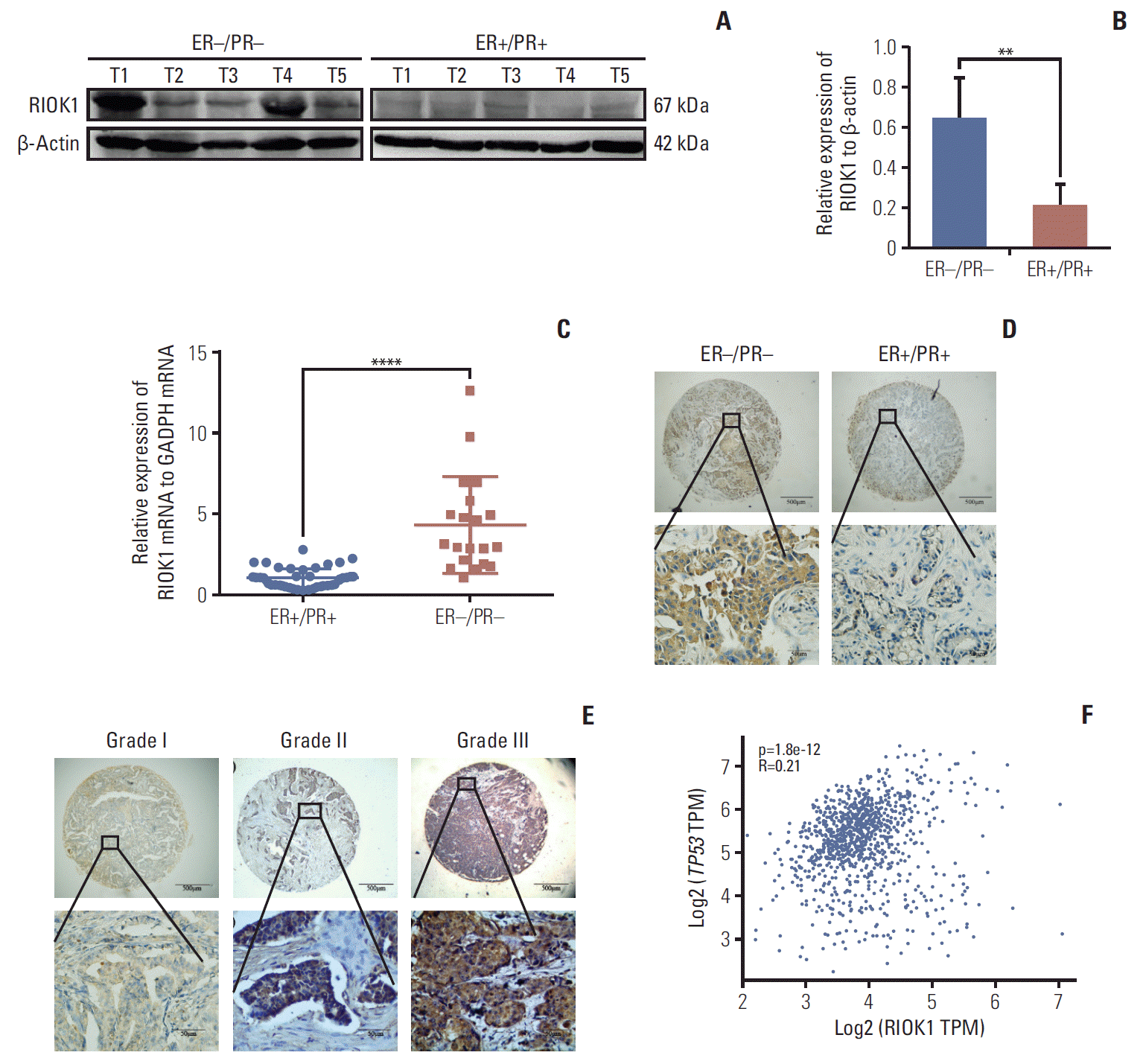
Fig. 2.
RIOK1 was significantly upregulated in hormone receptor (HR)–negative tissues. (A, B) Assessment of RIOK1 expression in five HR-positive and five HR-negative breast cancer (BC) tissue samples as analyzed via Western blot. (C) The expression of RIOK1 at the mRNA level was assessed in 40 HR-positive and 20 HR-negative BC tissues via quantitative real-time polymerase chain reaction. (D) Staining in HR-negative BC tissue samples (left) and in HR-positive BC tissue samples (right) in situ. Original magnification: ×40 (upper), ×400 (lower). (E) RIOK1 staining was weak in grade I tumors (left), moderate in grade II tumors (middle), and strong in grade III tumors (right). Original magnification: ×40 (upper), ×400 (lower). (F) The database shows the relevance between RIOK1 and p53. ER, estrogen receptor; PR, progesterone receptor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; TPM, transcripts per million. **p < 0.01, ****p < 0.0001 vs. HR-negative tissues.
![]()
Table 1.
The relationship between RIOK1 and p53 expression and clinicopathological parameters
| Clinicopathological parameter | No. |
RIOK1 |
p-value |
p53 |
p-value | ||
|---|---|---|---|---|---|---|---|
| Low or no | High | Low or no | High | ||||
| Total | 166 | 91 | 75 | 108 | 58 | ||
| Age (yr) | |||||||
| < 50 | 67 | 39 | 28 | 0.473 | 46 | 21 | 0.427 |
| ≥ 50 | 99 | 52 | 47 | 62 | 37 | ||
| Tumor diameter (mm) | |||||||
| ≤ 20 | 74 | 42 | 32 | 0.655 | 51 | 23 | 0.353 |
| > 20 | 92 | 49 | 43 | 53 | 75 | ||
| Tumor stage (TNM) | |||||||
| I | 52 | 29 | 23 | 0.269 | 38 | 14 | 0.077 |
| II | 86 | 51 | 35 | 55 | 31 | ||
| III-IV | 28 | 11 | 17 | 15 | 13 | ||
| Histological grade | |||||||
| I | 21 | 15 | 6 | 0.010* | 17 | 4 | 0.263 |
| II | 99 | 59 | 40 | 62 | 37 | ||
| III | 46 | 17 | 29 | 29 | 17 | ||
| Hormone receptor | |||||||
| ER | |||||||
| Negative | 65 | 24 | 41 | < 0.001*** | 35 | 30 | 0.015* |
| Positive | 101 | 67 | 34 | 73 | 28 | ||
| PR | |||||||
| Low | 74 | 27 | 47 | < 0.001*** | 39 | 35 | 0.003** |
| High | 92 | 64 | 28 | 69 | 23 | ||
| HR status | |||||||
| ER (+)/PR (+) | 89 | 62 | 27 | < 0.001*** | 66 | 23 | 0.003** |
| ER (–)/PR (+) | 3 | 2 | 1 | 3 | 0 | ||
| ER (+)/PR (–) | 12 | 5 | 7 | 7 | 5 | ||
| ER (–)/PR (–) | 62 | 22 | 40 | 32 | 30 | ||
| Molecular type | |||||||
| Luminal A | 27 | 17 | 10 | < 0.001*** | 23 | 4 | 0.012* |
| Luminal B | 74 | 52 | 22 | 51 | 23 | ||
| HER2+ | 38 | 13 | 25 | 18 | 20 | ||
| TNBC | 27 | 9 | 18 | 16 | 11 | ||
| Aggressive phenotype | |||||||
| HER2 | |||||||
| Low | 99 | 58 | 41 | 0.238 | 78 | 21 | < 0.001*** |
| High | 67 | 33 | 34 | 30 | 37 | ||
| Triple-negative phenotype | |||||||
| No | 141 | 82 | 59 | 0.040* | 93 | 48 | 0.568 |
| Yes | 25 | 9 | 16 | 15 | 10 | ||
| Lymph node metastasis | |||||||
| Negative | 92 | 54 | 38 | 0.263 | 54 | 38 | 0.263 |
| Positive | 74 | 37 | 37 | 37 | 37 | ||
| Ki67 | |||||||
| Low | 70 | 43 | 27 | 0.129 | 49 | 21 | 0.237 |
| High | 96 | 47 | 49 | 58 | 37 | ||
| Survival time (mo) | |||||||
| ≥ 80 | 128 | 78 | 50 | 0.011* | 88 | 40 | 0.228 |
| < 80 | 33 | 12 | 21 | 19 | 14 | ||
| p53 | |||||||
| Low | 115 | 65 | 40 | 0.008** | - | - | - |
| High | 51 | 20 | 31 | - | - | ||
![]()
3. Correlation between RIOK1 expression and prognosis in BC patients
We next further assessed how RIOK1 expression in BC tumor tissues was associated with BC patient survival outcomes using Kaplan-Meier survival analyses. Through this approach, we observed significantly longer overall survival (OS) in BC patients with low RIOK1 expression levels relative to those with higher expression of this protein (p=0.003) (Fig. 1D). This analysis further confirmed that molecular subtype was another key determinant of BC patient survival outcomes (p=0.017) (Fig. 1E). When comparing RIOK1-positive and -negative samples, a crude HR of 2.473 was calculated. A univariate analysis revealed that elevated RIOK1 expression (p=0.012), Ki67 levels (p=0.042), lymph node metastasis (p=0.001), and HER-2 positive (p=0.032) were all associated with a poorer BC patient prognosis (Table 2). A subsequent multivariate analysis further confirmed that both lymph node metastasis and RIOK1 expression levels were independent predictors of BC patient (p=0.003 and p=0.038, respectively) (Table 2).
Table 2.
Assessment of prognostic factors associated with BC patient 80-month survival via univariate and multivariate approaches
| Clinicopathological parameter |
Univariate analysis |
Multivariate analysis |
||
|---|---|---|---|---|
| Hazard ratio (95% CI) | p-value | Hazard ratio (95% CI) | p-value | |
| RIOK1 (high vs. low) | 2.473 (1.216-5.028) | 0.012* | 2.133 (1.041-4.372) | 0.038* |
| Ki67 (high vs. low) | 2.213 (1.028-4.763) | 0.042* | - | - |
| Lymph node metastasis (yes or no) | 3.364 (1.598-7.079) | 0.001** | 3.069 (1.449-6.498) | 0.003** |
| p53 (high vs. low) | 1.517 (0.749-3.071) | 0.247 | - | - |
| ER (yes or no) | 0.551 (0.278-1.090) | 0.086 | - | - |
| PR (yes or no) | 0.612 (0.308-1.215) | 0.160 | - | - |
| HER2 (yes or no) | 2.132 (1.068-4.255) | 0.031* | - | - |
| Age stage (≤ 50 yr vs. > 50 yr) | 1.471 (0.713-3.033) | 0.296 | - | - |
| Tumor diameter (≤ 2 cm vs. > 2 cm) | 1.852 (0.881-3.892) | 0.104 | - | - |
| Grade (well/moderately vs. poorly) | 2.530 (0.605-10.577) | 0.203 | - | - |
![]()
4. RIOK1 promotes migration, invasion, and angiogenesis in BC cells
As the expression of RIOK1 was higher in the MDA-MB-231 cell line relative to in MCF7 cells, we knocked down and overexpressed RIOK1 in these respective cell lines (Fig. 3A, S1 Fig.). Given that metastasis contributes to the low survival rate of BC patients, we evaluated the role of RIOK1 in BC migration and invasion. Transwell and wound healing analyses revealing that RIOK1 knockdown impaired cell motility, whereas RIOK1 overexpression had the opposite effect (Fig. 3B-D). Angiogenesis is an additional key driver of tumor metastasis. The tube formation assay revealed that when RIOK1 was knocked down, tube formation was impaired in MDA-MB-231 cells (Fig. 3E). Consistent with these data, the expression of E-cadherin was increased and vimentin and N-cadherin were markedly downregulated when RIOK1 was knocked down (Fig. 3F).
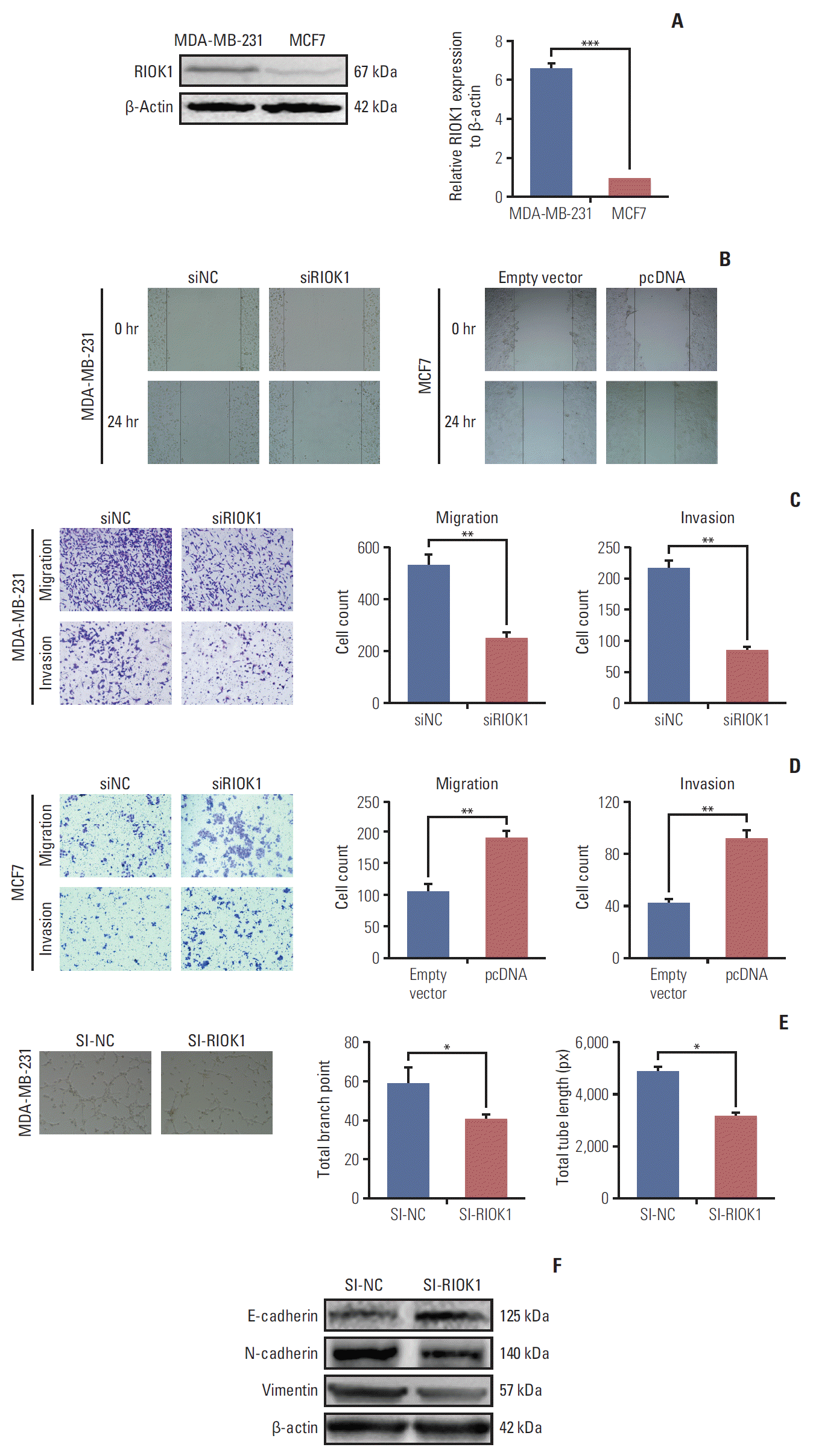
Fig. 3.
Inhibiting RIOK1 expression decreased breast cancer (BC) cells migration, invasion, and angiogenesis. (A) Relative expression of RIOK1 in BC cell lines. (B) A wound healing assay was performed to determine the migration of MDA-MB-231 and MCF7 cells. (C, D) Transwell assay was used to measure the capacity of invasion and migration capacity on BC cells transfected with siRIOK1 or pcDNA compared with siNC or empty vector. (E) The effect of the RIOK1 expression of MDA-MB-231 cells on endothelial cell tube formation. (F) Western blot analysis was used to detect the RIOK1 level on epithelial-mesenchymal transition process markers. *p < 0.05, **p < 0.01, ***p < 0.001.
![]()
5. RIOK1 modulates BC cell proliferation, cell cycle progression, and apoptosis
CCK-8 and colony formation assays indicated that RIOK1 affected the proliferation of BC cells (Fig. 4A-D). We additionally used EdU incorporation assays to assess the percentage of proliferating BC cells when RIOK1 was knocked down (Fig. 4E). Flow cytometry revealed that BC cells exhibited G2/M phase arrest when RIOK1 was knocked down (Fig. 4F), with these cells additionally exhibiting increased rates of apoptosis, while apoptosis was inhibited in MCF7 cells overexpressing RIOK1 (Fig. 4G and H). We then used western blot to measure the expression of proteins related to the above analyses. In light of their importance in HR-negative BC, we analyzed the PI3K/AKT and MAPK/ERK signaling pathways in MDA-MB-231 cells, revealing that phospho-AKT and phospho-ERK1/2 levels were downregulated when RIOK1 was knocked down, indicating that RIOK1 may affect BC cells via these two pathways (Fig. 4I).
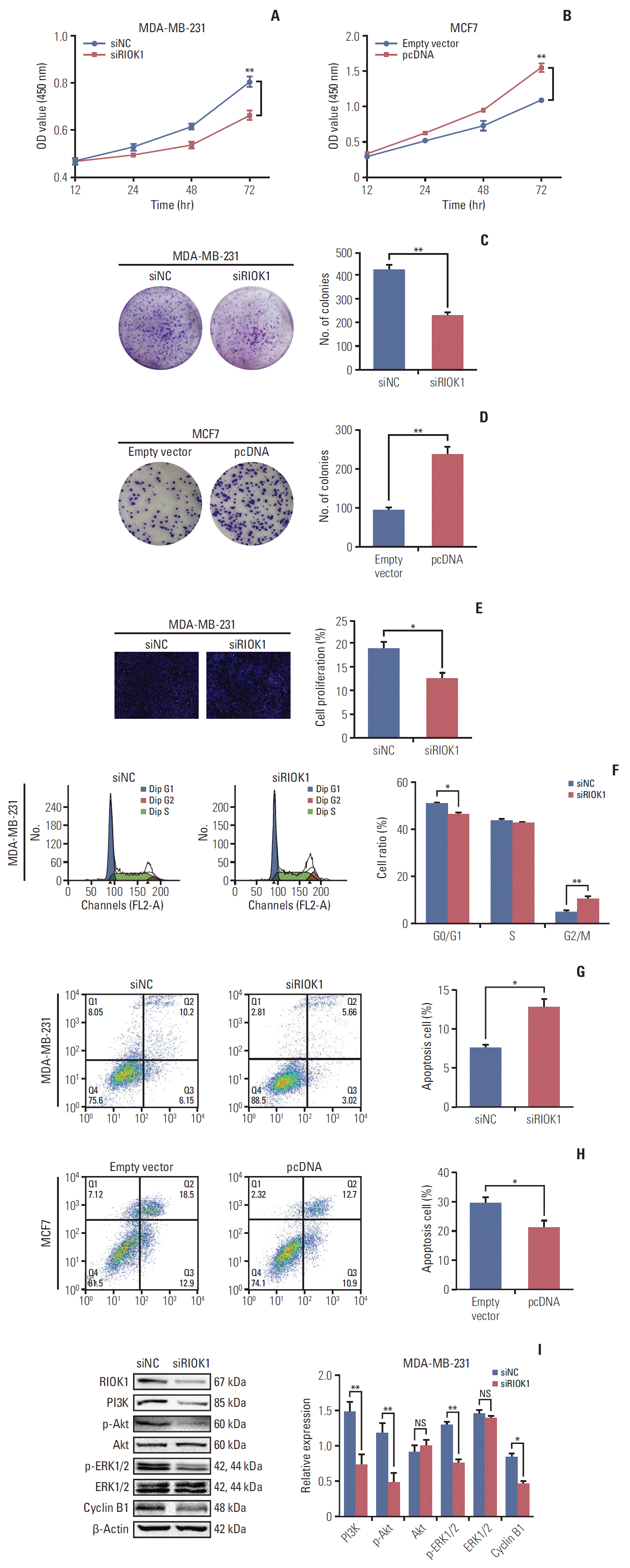
Fig. 4.
RIOK1 promoted breast cancer (BC) cells proliferation, induced cell cycle arrest, and reduced cell apoptosis. (A, B) Cell Counting Kit-8 assay presented the proliferation capacity of MDA-MB-231 and MCF7 cells transfected with siRIOK1 or pcDNA compared with siNC or empty vector. (C, D) Colony formation assay was used to detect the proliferation of MDA-MB-231 and MCF7 cells transfected with siRIOK1 or pcDNA. (E) EdU assay was used to evaluate cell proliferation. Representative images for EdU-positive cells (red) and Hoechst-stained nuclei (blue) of MDA-MB-231 transfected with siRIOK1 or siNC were shown in the left. Quantification data was shown in the right. (F) Flow cytometry of cell cycle was used to analyze the effect of siRIOK1 on MDA-MB-231 cells. (G, H) Induction of apoptosis of BC cells was analyzed by flow cytometry on MDA-MB-231 and MCF7 cells transfected with siRIOK1 or pcDNA compared with siNC or empty vertor. PI3K, phosphoinositide 3-kinase. (I) Western blot analysis of proliferation-related and apoptosis-related pathway proteins on MDA-MB-231 cells transfected with siRIOK1 or siNC. *p < 0.05, **p < 0.01.
![]()
6. Knockdown of RIOK1 inhibits tumor growth in vivo
We next investigated whether RIOK1 was able to modulate BC tumorigenesis in vivo. MDA-MB-231 cells transfected with siRIOK1 or siNC were injected into nude mice and tumor growth was then monitored, revealing that tumors were smaller in mice in the siRIOK1 group relative to the siNC group (Fig. 5A-D). We additionally confirmed via qRTPCR that RIOK1 expression was knocked down by siRIOK1 (Fig. 5E). These results thus suggested that knockdown of RIOK1 was able to inhibit tumor growth in vivo.
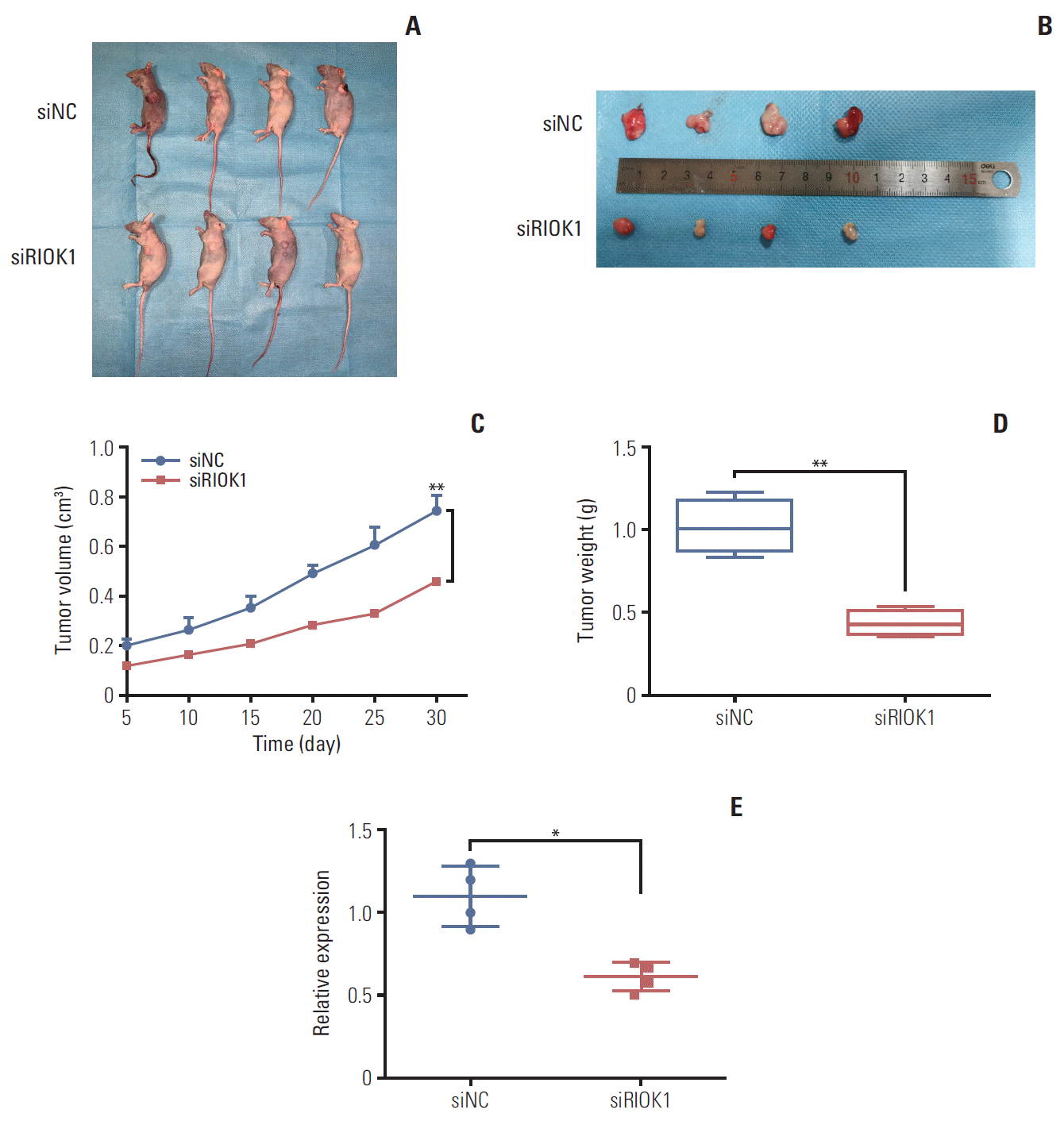
Fig. 5.
Knockdown of RIOK1 inhibited tumor growth in vivo. (A, B) Knockdown of RIOK1 inhibited the growth of MDA-MB-231 cells in nude mice. (C) Growth curves of xenograft tumors with siRIOK1 or siNC. (D) The weight of tumors from two groups after removal. (E) RIOK1 expression in tumor tissues was detected by quantitative real-time polymerase chain reaction. *p < 0.05, **p < 0.01.
![]()
7. miR-204-5p modulates RIOK1 expression and impact HR-negative BC
To identify potential miRNAs that target RIOK1, we used TargetScan, miRDB, starBase, and Tarbase to identify miR-204-5p as the only predicted miRNA targeting this gene (Fig. 6A). We then detected the expression of RIOK1 in MDA-231 cells which was transfected with a miR-204-5p mimic or inhibitor (Fig. 6B). A luciferase reporter assay confirmed that miR-204-5p and RIOK1 were able to directly interact with one another in cells (Fig. 6C). Furthermore, we performed rescue assays to validate whether miR-204-5p was able to reverse the pro-tumorigenic role of RIOK1 in BC cells, and we determined that miR-204-5p/RIOK1 promoted BC cell proliferation and migration in vitro (Fig. 6D-F). Furthermore, through a database search, we found that lower miR-204 expression was predictive of a worse outcome in BC patients (S2 Fig.). Together, these data suggested that miR-204-5p was able to impact RIOK1 and to modulate its ability to influence tumor progression.
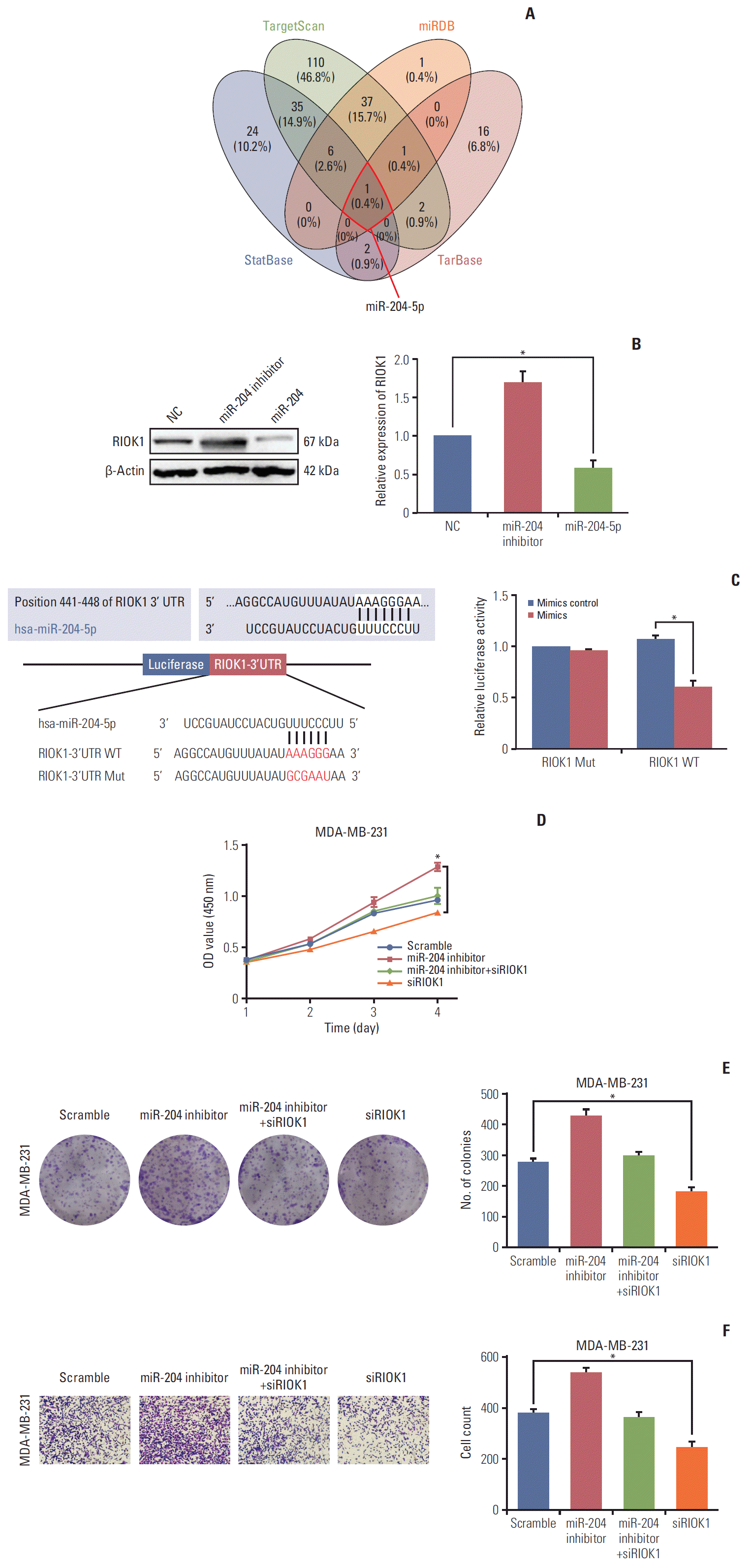
Fig. 6.
miR-204-5p directly targeted on RIOK1 and regulated the effects of RIOK1 on hormone receptor (HR)–negative breast cancer (BC) cells. (A) Bioinformatics analysis of predicted miRNAs interacted with RIOK1. (B) Western blot analysis of RIOK1 expression in MDA-MB-231 after transfection with miR-204-5p or inhibitor. (C) The luciferase reporter assay showed a relationship between miR-204-5p and RIOK1. (D, E) Effect of miR-204-5p and RIOK1 on BC cell proliferation capability. (F) Effect of miR-204-5p and RIOK1 on BC cell migration capability. *p < 0.05.
![]()
Discussion
RIOK1 is a key kinase in the RIO protein family. Kinases are essential regulators of all cellular processes, and make up roughly 2% of all eukaryotic genes [17], regulating transcription, translation, and protein function via phosphorylation of specific target proteins [18]. While best studied in yeast, RIO kinases are also thought to be key regulators of ribosomal biogenesis in mammalian cells. Indeed, RIOK1 has been shown to be closely linked to mammalian cell proliferation, with its absence leading to cell cycle arrest at the G2/M checkpoint in affected cells [19]. The dependent of cell proliferation on RIOK1 activity was evident even in a study of RAS-driven tumor cells, suggesting that therapeutic targeting of RIOK1 may be a viable strategy for treating such RAS mutant cancers [20]. RIOK1 has also been shown to be overexpressed in CRC and NSCLC [15], but it has not been directly studied in the context of BC.
BC remains the most common invasive cancer type affecting women [21], with these tumors being classified using a number of different systems that offer insight into the associated disease prognosis and amenability to therapeutic treatment. In the present study, for the first time, we detected high RIOK1 expression in approximately half of BC patient tumors, with this protein primarily localizing to the cytoplasm in these cancer cells. These findings suggested that RIOK1 may not undergo nuclear translocation, or that higher expression may be associated with increased cytoplasmic localization. We similarly found that the expression of RIOK1 at both the mRNA and protein level was significantly correlated with tumor HR status, tumor grade, and with patient survival outcomes. Specifically, HR-negative tumors exhibited higher RIOK1 expression, and such elevated RIOK1 expression was in turn associated with reduced BC patient OS in both univariate and multivariate analyses. These results, therefore, indicated that RIOK1 expression level is an independent predictor of BC patient survival outcomes. HR status has been regarded as a determining factor of prognosis and lacked of specific molecular therapy. We found the clinical association between RIOK1 and HR, however, the relationship between RIOK1 and HR was unclear, and further experiments are required to explore this association.
Interestingly, elevated RIOK1 expression was shown to be associated with elevated p53 levels in this analysis, and this has not been previously reported in other cancers. In addition, HR-negative BC patient samples exhibited elevated p53 levels in much the same way that they exhibited higher levels of RIOK1 expression. p53 is well known to function as a tumor suppressor gene, with the wildtype version of the p53 protein functioning to promote DNA repair and cell cycle arrest when it is [22,23]. Mutations that disrupt such wild type p53 functionality, however, are very common in tumors, affecting upwards of 80% of triple-negative BC and other serious tumor types [24]. As a result, such p53 mutations in immunohistochemistry are closely associated with reduced BC patient survival [25,26]. Previous work by Darb-Esfahani et al. [27] has further demonstrated that p53 levels are higher in triple-negative BC (74.8%) and HER2-positive BC (55.4%), suggesting that such tumors may express high levels of mutant p53 that are associated with a poorer patient prognosis. Previous studies have largely failed to address the relationship between RIO family proteins and p53, with just one study having demonstrated an interaction between these proteins and p53 activity [14]. In this study, only clarified the clinical link between RIOK1 and p53, but further follow-up experiments will be needed to understand the mechanistic basis for this association.
Finally, through cell phenotypes assays, we found RIOK1 could affected various tumor process such as proliferation, apoptosis, cell cycle, migration, and ivnvasion of BC cells. To investigate the mechanism on the subsequent studies, we detected several protein levels that regulate tumor process such as epithelial-mesenchymal transition, PI3K/AKT, and MAPK/ERK. To figure out what regulate RIOK1, we used four databases to find out the potential upstream miRNA. Only one miR-204-5p was in the list. MiR-204-5p was reported to be a tumor suppressor in BC patients and predicted poorer prognosis [28]. Shen et al. [29] revealed that the upregulation of miR-204-5p inhibits the proliferation, invasion, and migration of BC cells. In addition, in BC spheroids, miR-204 was downregulated in MDA-MB-231 cells than in MCF-7 cells, which means miR-204 could be associated with HR status, similarily [30]. We verified the relationship between miR-204 and RIOK1 by luciferase assay and co-transfected miR-204 and siRIOK1. According to our study, several assays indicated that miR-204-5p could regulate RIOK1 activity. Therefore, we speculated that miR-204-5p could be a potential regulator of RIOK1.
In summary, the results of this analysis for the first time indicate that HR-negative BC tumor tissues exhibit higher levels of RIOK1 expression, with RIOK1 expression also being associated with other clinicopathological characteristics in these patients. Importantly, elevated RIOK1 levels were associated with a poorer BC patient prognosis. RIOK1 expression levels were also associated with those of p53, which is already known to be a predictor of poor BC patient outcomes. RIOK1 could regulate BC cells proliferation, apoptosis, and metastasis by affecting the PI3K/AKT and MAPK/ERK signaling pathways in HR-negative BC cells. As such, we identified RIOK1 as a potential therapeutic target in patients with BC, although further in-depth multi-disciplinary studies will be needed to validate this possibility.
 XML Download
XML Download