

 PDF
PDF Citation
Citation Print
Print


Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor accounting for up to 6%-10% of all childhood cancers and it is one of the leading cause of cancer mortality in children [1]. NB arises from the precursor cells of the sympathetic nervous system or the adrenal medulla. Since NB is widely heterogeneous in its clinical phenotypes and treatment outcomes, current treatment protocols are based on risk stratification of NB. High-risk group is currently defined as MYCN-amplified tumors at any age or metastatic tumors in patients older than 18 months according to the International Neuroblastoma Risk Group (INRG) classification system [2]. The patients in the high-risk group show poor prognosis despite modern intensive multimodal treatment. N-myc proto-oncogene protein, also known as N-Myc, is encoded by the MYCN gene in humans. Since MYCN amplification was found to be a highly predictive marker of poor outcome in the NB patients by the INRG cohort, the MYCN status is used for risk stratification [3]. Based on this, approximately 40% of NB patients have been classified as high-risk [2,4]. In a previous study regarding familial NB, PHOX2A, ALK, KIF1Bβ, and RAS mutations were reported as causal mutations [5]. However, while only 1%-2% of NB cases were familial, further genetic association studies are required to identify the predisposing genetic factors. In 2008, the first genome-wide association study (GWAS) was performed for Europeans in a case of sporadic NB [6]. They reported three variants of chromosome 6p22, which have been mapped to the genes CASC-15 and NBAT-1. Several candidate genes such as LMO1, BARD1, HACE1, and LIN28B have been reported by subsequent GWAS [5]. Capasso et al. [7] investigated the genetic factor for the NB patients who developed high-risk tumors and they found that the locus in 6p22 was enriched. Additionally, they found several novel risk-related single nucleotide polymorphisms (SNPs) including intronic variant in BARD1 gene [7]. Although previous GWAS and candidate genetic studies have provided considerable information about the genetics and understanding for NB, most of these studies were performed on Caucasian and African Americans. To expand the genetic studies for other various populations such as Asians, it is necessary to explain the genetic aspects of NB.
Due to the remarkable phenotypic heterogeneity of NB, the mild to moderate effects on the risk of NB development are still unclear. In this study, we have performed GWAS using NB to discover genetic variants in Korean children. To our knowledge, this is the first GWAS study in Korean NB patients.
Materials and Methods
1. Study subjects
We screened patients who were diagnosed with NB between February 1998 and March 2017. After screening, 296 NB patients whose peripheral blood samples were already cryopreserved at Samsung Medical Center Biobank were enrolled in this study. The 77,472 exome chip genotypes of the healthy controls (n=1,000), without any history of tumor, were obtained from the National Biobank of Korea (No. 2018-019). Medical records were reviewed for obtaining detailed clinical and biological data such as the clinical features presented during diagnosis, tumor biology including MYCN amplification status and tumor histology by International Neuroblastoma Pathology Classification (INPC). During the study period, the patients were classified into high-risk group and non–high-risk group according to their age during diagnosis, tumor stage based on the International Neuroblastoma Staging System (INSS), and MYCN amplification status. In brief, stage 4 tumors in patients older than 1.5 years or with MYCN-amplified tumors were included in the high-risk group.
2. Genome-wide genotyping
Genomic DNA was extracted from the peripheral blood lymphocytes of the patients using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI), according to the manufacturer’s protocol. Approximately 200 ng of genomic DNA was used to genotype each sample using the Illumina’s Global Screening Array (GSA) BeadChip (Illumina, San Diego, CA). The samples were then processed according to the Illumina Infinium assay manual. Each sample was wholegenome amplified, fragmented, precipitated, and resuspended into an appropriate hybridization buffer. The denatured samples were then hybridized on a prepared GSA BeadChip for a minimum of 16 hours at 48°C. Following hybridization, the BeadChips were processed for the single-base extension reaction, staining, and imaging on an Illumina iScan system. The normalized bead intensity data obtained for each sample were uploaded onto the GenomeStudio software (Illumina) which converted the fluorescent intensities into SNP genotypes. The quality of sample was checked by sample call rate (> 95%). The quality of cluster for marker was measured by GenTrain scores, and then high-quality markers used this study (> 0.7).
3. Imputation and statistical analysis
The clinical variables were summarized using mean±standard deviation or median (range), as appropriate (Table 1). We performed imputation consisting of 1,296 samples. The prephasing of genotypes was conducted with SHAPEIT.v2.r837. We then imputed variants from the 1000 Genomes Project phase 3 references using IMPUTE v2.3.2. Markers with low imputation quality as call rate (< 98%), minor allele frequency (MAF < 1%), p-value of Hardy-Weinberg equilibrium (HWE; < 1E-5), duplicated markers, and ambiguous strand markers were excluded from the association analysis. In addition, low-quality samples (call rate < 95%) were applied for quality control. For genome-wide association analysis, genotype distributions were compared using logistic regression analyses with the HelixTree software (Golden Helix Inc., Bozeman, MT). To predict a protein damaging score for each nonsynonymous SNP, PolyPhen-2 program [8] was used (http://genetics.bwh.harvard.edu/pph2/index.shtml) according to the manual. Gene pathway analysis for significantly associated SNPs with the risk of NB was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) functional annotation tool (https://david.ncifcrf.gov/). In addition, biological network analysis was performed by GluGo, Cytoscape plug-in that visualizes non-redundant biological terms for large clusters of genes in a functionally grouped network (https://cytoscape.org/).
Results
1. Clinical characteristics
A total of 296 NB patients were recruited for the current study. In addition, genotypes of 1,000 normal healthy controls were obtained from the National Biobank of Korea. The average age of the NB patients was 2.1 years (range, 0.0 to 19.3 years). Among the 296 patients, 142 patients were stratified into high-risk group and MYCN amplification was seen in 56 patients. Table 1 shows the characteristics of the patients and healthy controls.
2. Association analysis and identification of novel susceptibility loci
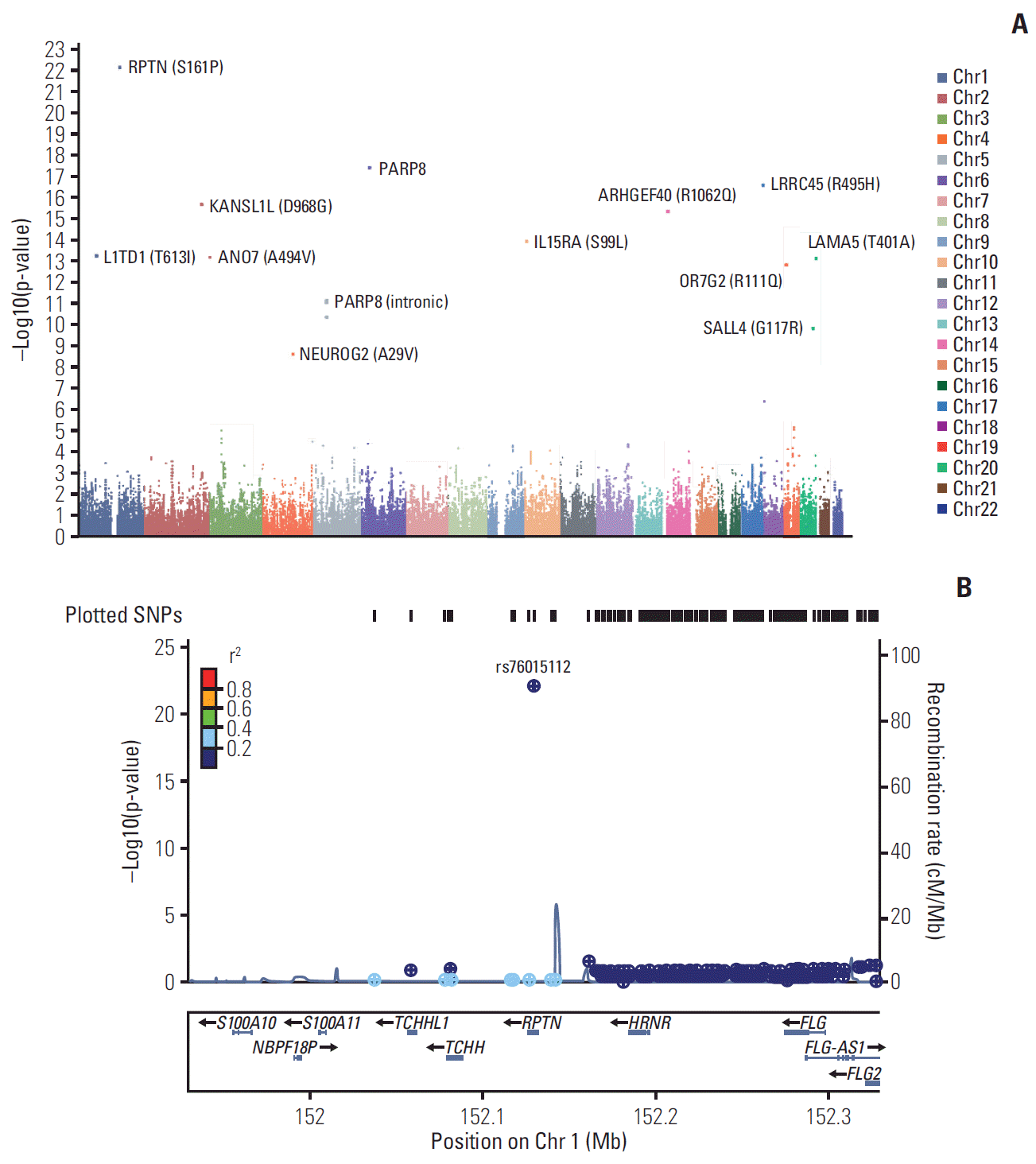
A quantile-quantile plot for the association test between NB and healthy controls showed a significant deviation of measures at the tail (S1 Fig.) indicating potentially true associations between the SNPs and NB. A total of 281K markers were imputed from 535K genotypes of patients and 76K genotypes of healthy controls using strict quality control parameters (MAF > 1%, missing rate < 1%, or p for HWE < 1×10–5). First, we tested the association between NB patients and healthy controls using logistic regression analysis. A total of 21 markers showed significant association with the risk of NB after adjusting for multiple comparisons (pcorr < 0.05) (Fig. 1A). The significant markers for the risk of NB are summarized in Table 2. The markers were located in RPTN, MRPS18B, LRRC45, KANSL1L, ARHGEF40, IL15RA, L1TD1, ANO7, LAMA5, OR7G2, SALL4, and NEUROG2 genes. In addition, PARP8, EPB41L3, and MAP4K1 genes were found to be the nearby genes for the markers such as rs7717033, rs1375128, rs10737958, rs2594708, rs2463796, rs35296988, rs3864235, rs32396, rs9964022, rs17847695, rs117910631, rs147260795, rs146801912, rs17847686, rs200216392, rs77270842, rs149013375, and rs74990833 (Table 2). Among all the markers, 12 markers were distributed in the coding region. Most of the nonsynonymous SNPs showed low MAF except of rs76015112 (MAF < 0.010). In rs76015112, the MAF of NB was lower than healthy controls (0.125 vs. 0.332) as shown in Table 2. However, the other markers in the coding region showed a high MAF in NB compared to the controls (Table 2). We analyzed the regional association of 400 kb around RPTN on chromosome 1q21.3 (Fig. 1B) and observed that the rs76015112 marker showed relatively robust association signal (pcorr=2.3E-17) (Table 2). The results of linkage disequilibrium (LD) analysis showed that the marker was unlikely to be in LD with the nearby genes (Fig. 1B).
3. Association analysis of NB subgroups
The NB patients were classified as either with MYCN amplification or without. Table 3 shows the significant SNPs after genome-wide association analysis between MYCN-amplified NB patients and the other NB patients. Interestingly, many SNPs within ADGRE1 (synonyms: EMR1) gene showed significant associations (S2A Fig.). Among them, rs725614 and rs457857 had nonsynonymous SNPs as V589I (NM_001256253) and I424V (NM_001256253) (Table 3). In the regional association analysis of 200 kb around ADGRE1 (synonyms: EMR1), we found that 32 SNPs were in tight LD (S2B Fig.). In addition, three significantly associated SNPs were found to be located in the intron region of C2CD6 gene (rs143074421, rs77468686, and rs117943473).
In the following analysis, GWAS was performed with NB patients in high-risk group and non–high-risk group. The top 30 significant SNPs are listed in Table 4. As a result, rs62296061 located intron region in FGFGL1 gene was found to be highly associated with the high-risk group. Many SNPs within ADGRE1 gene showed significant associations (S3A Fig.). In the rs725614 and rs457857 located in coding region, the MAF in high-risk group was lower than other groups (0.080 vs. 0.217 in rs725614, 0.109 vs. 0.233 in rs457857). In the regional association analysis of 200 kb around ADGRE1, 32 SNPs were found to be in tight LD (S3B Fig.). In Table 4, we have shown that PON1 and PON2 genes were located near seven markers such as rs11981667, rs17166829, rs73422040, rs11980347, rs17884252, rs17883750, and rs149643570.
4. Assessment of gene functional annotation and biological network analyses
The result of Gene Ontology (GO) analysis was listed in S4 Table. A total of 37 significant GO terms were identified. Among them, 10 biological pathways such as lipid transporter activity, kidney morphogenesis, regulation of macrophage derived foam cell differentiation, positive regulation of macrophage derived foam cell differentiation, sensory perception of taste, negative regulation of cell division, foam cell differentiation, macrophage derived foam cell differentiation, dicarboxylic acid catabolic process, and synaptic membrane adhesion was maintained the signals after multiple correction (pcorr < 0.05). S5 Fig. shows the result of biological network related to other biological functions. In the regulation of macrophage derived foam cell differentiation, it was closely linked to positive regulation of macrophage derived foam cell differentiation. To examine the biological function, we performed GO analysis using the DAVID. We have shown the result of GO analysis for the identified significant SNPs using the risk of NB using DAVID in S4 Table. Ten GO terms were observed have significantly corrected p-value (< 0.05). These were lipid transporter activity, kidney morphogenesis, regulation of macrophage derived foam cell differentiation, positive regulation of macrophage derived foam cell differentiation, sensory perception of taste, negative regulation of cell division, foam cell differentiation, macrophage derived foam cell differentiation, dicarboxylic acid catabolic process, and synaptic membrane adhesion.
Discussion
In the current study, we investigated novel genetic susceptibility markers for the risk of NB and its subgroups such as MYCN amplification group and high-risk group. We used the imputed markers from the genotypes of Illumina’s GSA BeadChip through strict quality control. The Illumina GSA BeadChip, which was recently launched, contains highly optimized multi-ethnic clinical markers from well-defined databases of known diseases such as ClinVar, the Pharmacogenomics Knowledgebase (PharmGKB), and the National Human Genome Research Institute (NHGRI)-EBI database.
According to the INRG, four categories (very low-risk, low-risk, intermediate-risk, and high-risk) could be classified by seven clinical and biological factors [4]. Over the past 10 years, several genetic studies have been performed to identify somatic and germline variants affecting the onset and survival rate of NB. In the first GWAS, CASC-15 and NBAT-1 genes found on chromosome 6p22 were identified as the susceptible genes. Interestingly, we identified significant markers in cases with high-risk or MYCN-amplified NB. Subsequently, it was found that rs6939340 was most significantly associated with the risk of sporadic NB [6]. In the next GWAS, several novel risk SNPs including BARD1 gene on chromosome 2q35 were identified [7]. Other GWAS conducted using in familial and sporadic cases of NB have reported novel additional risk SNPs in candidate genes such as DUSP12, HSD17B12, DDX4, IL31RA, LMO1, HACE1, LIN28B, SPAG16, NEFL, TP53, CPZ, MLFL, CDKN1B, KIF15, and MMP20 [9-19]. Among them, SNPs in BARD1 and LMO1 candidate genes were replicated in other cohorts due to its association by large cohort study [19,20]. In the current study, we also identified significant associations between the genes and the risk of NB (p=0.001, rs3768716, rs2070094 in BARD1 gene; p=0.0002, rs110419 in LMO1 gene).
However, as the previous GWAS studies were performed mostly with Caucasian and Africans, there is a serious lack of GWAS studies for Asian populations. In the current study, we identified novel genetic markers for comparing the risk between children with NB and healthy controls with no tumor record in Korean population. Using high-quality markers and strict criteria, imputed markers were used. As a result, we found 21 statistically significant markers associated with the risk of NB (pcorr < 0.05) such as RPTN, MRPS18B, LRRC45, KANSL1L, ARHGEF40, IL15RA, L1TD1, ANO7, LAMA5, OR7-G2, SALL4, and NEUROG2. Interestingly, out of these 21 significant SNPs (pcorr < 0.05), 12 SNPs were nonsynonymous (average MAF=0.064). Except rs76015112, rs77226427, and rs2886644, most nonsynonymous SNPs showed rare MAF above 5%. The most significantly associated marker was found to be rs76015112 which was located on the RPTN gene (p=8.1E-23, pcorr=2.3E-17). The RPTN gene encodes for repetin, an extracellular epidermal matrix protein consisting of 784 amino acids. This protein is rich in glutamine with EF-hands of the S100 type and contributes to the formation of the cornified envelope [21]. However, this gene has not yet been reported to be associated with NB. Interestingly, three nonsynonymous SNPs (rs117249618, rs117674897, and rs114591848) in LRRC45, KANSL1L, and ARHGEF40 genes showed high protein function damaging scores (1.00, 0.88, and 0.99). Moreover, the MAF of the SNPs was higher in NB than in controls (Table 2). This implies that the SNPs may be risk factors for the onset of NB. Candidate markers within BAL1, LMO1, MLF1, and HACE1 previously reported to be related with the risk of NB in Caucasian and European were not replicated in this study. The reason is presumed to be due to ethnic difference and platform difference.
NB patients were further divided into two subgroups, MYCN-amplified group and high-risk group, and then logistic regression analysis was performed for each of the subgroups. In these two subgroups, we found that many significant SNPs were located in the ADGRE1 gene. Interestingly, two nonsynonymous SNPs (rs7256147 and rs457857) were identified in the ADGRE1 gene (Tables 3 and 4). The frequencies of the two nonsynonymous SNPs in MYCN-amplified or high-risk tumors were lower than those in MYCN non-amplified or non-high-risk tumors, respectively. This implies that the SNPs might have a protective role against the risk of NB. ADGRE1 gene has been renamed as EMR1 gene, which encodes for epidermal growth factor–like module containing mucin-like hormone receptor 1. ADGRE1 gene encodes for proteins belonging to a group of hormone receptor with seven transmembrane segments [22]. Therefore, the mutated product of the ADGRE1 gene might act as a neurotransmitter and influence the biological function of the signal transduction. When calculated using the GAS Power Calculator (http://csg.sph.umich.edu/abecasis/cats/gas_power_calculator), the expected power was 0.449. This result suggests that this study used insufficient samples, and that further studies using a large number of samples through multi-center collaboration in Asia are needed to verify the significant markers found by GWAS analysis.
To our knowledge, this is the first GWAS study in Korean NB patients. We discovered novel susceptible SNPs for the risk of NB. Of them, 12 nonsynonymous SNPs were identified. When the protein damaging prediction was performed by PolyPhen-2 algorithm, three SNPs showed high protein activity damaging scores (> 0.8). Additionally, we performed GWAS for two subgroups, MYCN-amplified group and the high-risk group, and identified the significantly associated SNPs in the ADGRE1 gene. The identified variations may helpful to investigate the potential function for the onset of NB by functional assay. Our study may provide a new direction in the formulation of medication for the risk of NB in Korean population.
 XML Download
XML Download