

 PDF
PDF Citation
Citation Print
Print


Introduction
Breast cancer is one of the most common malignant cancers among women worldwide with more than 2 million new cases expected to be diagnosed and 600 thousand deaths in 2018 [1]. Although current treatments, to a certain extent, effectively reduce tumor bulk, many patients with breast cancer still experience recurrence, metastasis, and ultimately death.
Cancer stem cells (CSCs) are a small subset of poorly differentiated tumor cells within a tumor that show self-renewal and differentiation into heterogeneous lineages [2]. CSCs also account for chemo- and radioresistance, resulting in tumor recurrence and eventually metastasis due to the failure of current treatments [2]. Moreover, breast cancer stem cells (BCSCs) are regulated by several transcriptional factors and signaling pathways, such as Nanog, Oct4, Notch pathway, and Wnt/β-catenin pathway [3]. Extensive evidence has shown that BCSCs are a crucial factor that contributes to the initiation, development, relapse, and chemoresistance of breast cancer.
A growing body of evidence supports aberrant glycosylation of cell surface receptors as a consequence of oncogenic transformation in different aspects of tumor progression, including proliferation, invasion, angiogenesis, and metastasis [4]. Within a tumor, altered glycosylation of proteins is frequently attributed to abnormal expression of glycosyltransferases [5]. Therefore, these enzymes are considered therapeutic targets, as aberrant glycans may be involved in promoting tumor progression and metastasis.
The β1,4-galactosyltransferase (B4GalT) family belongs to type II membrane-bound glycoproteins mainly located in the Golgi apparatus that exclusively transfer an active UDP-galactose in a β1,4 linkage to acceptor sugars [6]. There are currently seven members of the β4GalT gene family, B4Gal-T1-T7, which participate in the biosynthesis of different glycoconjugates and saccharide structures with slightly different substrate affinities and end products [6]. Specifically, B4GalT5 has been reported to catalyze the addition of galactose to GlcNAcβ1-4Man of glycans and functions as an important regulator in extraembryonic development [6]. Considerable research has revealed that extraembryonic development shares similarities with tumorigenesis in terms of biological behaviors (such as migration and invasion, gene expression and protein profiles, signaling pathways, and cell differentiation) [7], strongly suggesting that B4GalT5 may be involved in modulating the stemness of BCSCs. In this study, we demonstrated that B4GalT5 is upregulated in BCSCs to maintain the stemness of these cells by glycosylating the receptor Frizzled-1 and constantly activating Wnt/β-catenin signaling. Furthermore, we showed that B4GalT5 is located on the cell surface of a small subset of breast cancer cells, but cell surface localization did not play a decisive role in the stemness of BCSCs.
Materials and Methods
1. Reagents
Roswell Park Memorial Institute (RPMI) 1640 medium, minimum essential medium (MEM), Leibovitz’s L-15 medium, Dulbecco’s modified Eagle medium: nutrient mixture F-12 (DMEM/F-12), epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), and B27 were purchased from Gibco (Grand Island, NY). Fetal bovine serum (FBS) and trypsin were purchased from Gibco-Invitrogen. Insulin and fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibody were purchased from Solarbio (Beijing, China). Antibodies to detect aldehyde dehydrogenase 1A1 (ALDH1A1), phosphorylated GSK3β (Ser 9), β-catenin, phosphorylated β-catenin (Ser 45), Erk, and His were products of Cell Signaling Technology (Boston, MA). SuperSignal West Femto Maximum Sensitivity Substrate, EZ-Link Sulfo-NHS-SSBiotin, Pierce NeutrAvidin agarose, anti-B4GalT5, FITC-conjugated anti-CD44 and allophycocyanin (APC)-conjugated anti-CD24 antibodies were obtained from Thermo Scientific (Waltham, MA). Anti–Frizzled-1 and phycoerythrin (PE)-conjugated anti-His antibodies were purchased from R&D Systems (Minneapolis, MN). PE-conjugated anti–epithelial specific antigen antibody was purchased from Biolegend (San Diego, CA). The primary antibodies (β-actin and glyceraldehyde 3-phosphate dehydrogenase) and the secondary antibodies were purchased from HuaBio (Hangzhou, China). Horseradish peroxidase (HRP)-streptavidin, biotinylated Ricinus communis agglutinin I (RCA-I), and agarose-bound RCA-I were obtained from Vector Laboratories (Burlingame, CA). 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St. Louis, MO). The ALDEFLUOR Kit was purchased from Stem Cell Technologies (Vancouver, BC, Canada). The Mycoplasma PCR Detection Kit, DAPI, cell lysis buffer for western blot and immunoprecipitation (IP), phenylmethanesulfonylfluoride (PMSF), MG132, membrane and cytosol protein extraction kit and BCA Protein Assay Kit were purchased from Beyotime Institute of Biotechnology (Shanghai, China). B4GalT5 siRNA, NC siRNA, shB4GalT5 plasmid, and shNC plasmid were constructed by GenePharma (Shanghai, China). Triptolide with purity > 99% was obtained from Shanghai Institute of Materia Medica. Wnt 3β and leupeptin were obtained from the Laboratory of Molecular Medicine at Ocean University of China.
2. Tissue microarray
Tissue microarray (TMA; HBreD090CS01) was purchased from Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). The TMA has 90 cores from 45 patients with invasive breast cancer, including 45 tumor tissues and 45 corresponding adjacent tissues. The immunohistochemical staining rate was classified as 0 (negative), 1 (1%-25% positive tumor cells), 2 (26%-50%), 3 (51%-75%), and 4 (76%-100%). Staining intensity was classified as 0 (absence of stained cells), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). The immunohistochemistry (IHC) score was calculated by multiplying the staining rate and intensity. Correlations were determined by Spearman’s coefficient of correlation.
3. Cell culture
MCF-7, adriamycin-resistant MCF-7 (MCF-7ADR), and MDA-MB-231 cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cell lines were validated using short tandem repeat analysis by Genesky Biotechnology Inc., Shanghai (Shanghai, China), and tested for the absence of mycoplasma contamination by PCR using the Mycoplasma PCR Detection Kit. MCF-7 cells were maintained in MEM supplemented with 10% FBS, 0.01 mg/mL human recombinant insulin, and 1 μM nonessential amino acids. MCF-7ADR cells were cultured in RPMI-1640 medium supplemented with 10% FBS. MDA-MB-231 cells were cultured in L-15 medium supplemented with 15% FBS. Adriamycin was added on time to maintain the drug resistance phenotype of MCF-7ADR cells.
4. MTT assay
The MTT assay was used to measure the inhibitory effect of compounds on the viability of cancer cells. Adherent cells were seeded in 96-well plates at a density of 5,000 cells per well. After 24 hours, the cells were treated with different concentrations of triptolide for 72 hours. Twenty microliters of MTT solution was added to each well and incubated for 4 hours at 37°C. Then, dimethyl sulfoxide was added to the wells and incubated overnight at 37°C. The absorbance at 570 nm was measured using a microplate reader (BioTek, Winooski, VT).
5. Clinical dataset analysis
To analyze the expression of B4GalTs in invasive breast carcinomas compared with normal breast tissues, we used The Cancer Genome Atlas (TCGA) breast dataset from the Oncomine browser (https://www.oncomine.org).
Kaplan-Meier plotter (http://kmplot.com/analysis/index.php?p=background) was used to analyze the correlation between B4GalT5 expression and recurrence-free survival (RFS) in patients with breast cancer in 120 months [8].
The coexpression of B4GalT5 and CCR7, C-X-C chemokine receptor 4 (CXCR4), and ATP binding cassette subfamily B member 1 (ABCB1) was assessed in invasive breast invasive carcinoma samples from TCGA dataset by ‘R2: Genomics Analysis and Visualization Platform’ (http://r2.amc.nl). We also analyzed the coexpression of B4GalTs and ALDH1A1 and c-myc from GEPIA 2 from Zhang’s lab [9]. Correlations were determined by Pearson’s coefficient of correlation.
6. Flow cytometry analysis and sorting
For analysis of BCSC and non-BCSC cell fractions, cells were incubated with FITC-conjugated anti-CD44 antibody and PE or APC-conjugated anti-CD24 antibody in phosphate buffered saline (PBS) containing 2% FBS at 4℃ for 30 minutes. BCSCs (CD44+CD24–/low) and non-BCSCs (CD44+CD24+) were analyzed and sorted by flow cytometry (MFLO XDP, Beckman Coulter, Pasadena, CA). The ALDEFLUOR Kit was used for the identification of BCSCs that express high levels of the enzyme aldehyde dehydrogenase (ALDH) according to the manufacturer’s protocol.
For analysis of the proportions of the CD24–/low cell population in the His+ cell population and His– cell population, cells were incubated with PE-conjugated anti-His antibody and APC-conjugated anti-CD24 antibody in PBS containing 2% FBS at 4°C for 30 minutes. The samples were analyzed by flow cytometry (MFLO XDP, Beckman Coulter). The calculation method is as follows.
The proportion of CD24–/low cells in the His+ cell population=His+CD24–/low cells %/His+ cells %.
The proportion of CD24–/low cells in the His– cell population=His– CD24–/low cells %/His– cells %.
7. Plasmids
The DNA fragment for B4GalT5 was cloned from human cDNA using the corresponding primers and inserted into the pLVX-IRES-ZsGreen1 vector. The DNA fragment for B4GalT5-His was cloned from human cDNA using primers including His sequence and inserted into the pBABE-puro vector. These constructed vectors were transformed into DH5α cells and then amplified. The primers for B4GalT5 were 5’-CCGCTCGAGTGGCTGCAGCATGCGCG-3’ and 5’-CGCGGATCCTCAGTACTCGTTCACCTGAG-3’. The primers for B4GalT5-His were 5’-CGCGGATCCATGCGCGCCCGCCGGGGGCTGCT-3’ and 5’-CCGGAATTCTCAATGGTGATGGTGATGATGGT-3’.
8. siRNA and plasmid transfections
For siRNA transfection, cells were transfected at 80% confluency in 6-well plates with 50 nM control siRNA or B4GalT5-targeting siRNA (si-B4GalT5) (sense, 5’-CGGAGUGAGUGGCUUAACAdTdT-3’; antisense, 5’-UGUUAAGCCACUCACUCCGdTdT-3’) using Lipo2000 transfection reagents according to the manufacturer’s instructions. The knockdown efficiency was examined by western blotting after 48 hours.
For plasmid transfection, cells were transfected with the plasmids using Lipo3000 transfection reagents according to the manufacturer’s instructions. After 48 hours, the cells were treated with G418 or puromycin to screen for stably transfected MCF-7ADR cell lines. The stably transfected MCF-7ADR cell lines are as follows: B4GalT5-knockdown MCF-7ADR (MCF-7ADR/shB4GalT5), B4GalT5-overexpressing MCF-7ADR (MCF-7ADR/oe-B4GalT5), and MCF-7ADR expressing B4GalT5-His (MCF-7ADR/B4GalT5-His).
9. Mammosphere formation assay
For analysis of the effect of B4GalT5 on mammosphere formation, a single-cell suspension was quantified and seeded in ultralow attachment 6-well plates at a density of 2,000 cells per well at 37℃ and 5% CO2. Mammospheres were formed in serum-free DMEM/F-12 medium containing 20 ng/mL EGF, 10 ng/mL bFGF, 2% B27, and 5 μg/mL insulin. After 7 days, mammospheres with a diameter of > 50 μm were counted using a cell imaging multi-mode reader (BioTek).
For determination of the effect of triptolide on mammospheres, single-cell suspensions were quantified and seeded in ultralow attachment 6-well plates at a density of 2,000 cells per well at 37℃ and 5% CO2. Mammospheres were formed in serum-free DMEM/F-12 medium containing 20 ng/mL EGF, 10 ng/mL bFGF, 2% B27, and 5 μg/mL insulin with treatment with PBS or different concentrations of triptolide. After 7 days, mammospheres with a diameter of > 50 μm were counted using a cell imaging multi-mode reader (BioTek).
10. Colony formation assay
Five hundred microliters of pre-warmed 2×1,640 medium containing 20% FBS, 200 U/mL penicillin, 200 μg/mL streptomycin, and 500 μL of melted 1.2% agarose solution was mixed and transferred to a 6-well plate. When the bottom agar layer solidified, 1 mL of melted 0.7% agarose solution and 1 mL of single-cell suspension at a density of 500 cells per mL treated with increasing concentrations of triptolide were mixed and seeded on the bottom agar layer. The cells were incubated for 30 days at 37℃ and 5% CO2. The colonies were treated with 0.5 mL of MTT per well for 30 minutes at 37℃, and colonies containing up to 50 cells were counted.
11. Protein extraction, pull down, and immunoblotting
For the RCA-I pull-down assay, cells were lysed on ice for 30 minutes in cell lysis buffer for western and IP with 1 mM PMSF. Three hundred micrograms of total cell lysates were centrifuged at 12,000 ×g and 4°C for 10 minutes and then incubated with RCA-I agarose beads at 4°C overnight. After the samples were washed three times using cell lysis buffer, pulled-down proteins were subjected to western blotting. Total Frizzled-1 expression level was used as loading control.
For western blotting, membrane and cytosol proteins of MCF-7ADR cells were extracted by the membrane and cytosol extraction kit according to the manufacturer’s instructions. Total cell lysates were prepared using loading buffer on ice for 45 minutes and denatured at 95℃ for 15 minutes followed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Then, the samples were transferred to Immobilon-P polyvinylidene difluoride membranes, blocked with 5% non-fat milk, and incubated with the indicated antibodies. SuperSignal West Femto Maximum Sensitivity Substrate was used to detect HRP-conjugated secondary antibodies.
12. Cell surface biotinylation assay
After the samples were washed twice with cold PBS, MCF-7ADR cells were incubated with EZ-Link Sulfo-NHS-SSBiotin at a final concentration of 0.5 mg/mL for 60 minutes at 4°C, followed by a 50 mM glycine/PBS wash and two washes with PBS. Biotinylated cells were lysed with cell lysis buffer for western and IP and then centrifuged for 15 minutes at 12,000 ×g and 4°C. The supernatant was incubated with PNeutrAvidin agarose beads and mixed at 4°C overnight. Biotinylated proteins were eluted from the beads by heating to 100°C for 5 minutes in SDS-PAGE sample buffer before loading into the SDS-PAGE gel for western blotting.
13. Immunofluorescence assay
For detection of the p-GSK3β (Ser 9) and B4GalT5 expression levels, cells were cultured on a 96-well black-wall glass bottom plate for 24 hours. They were fixed with 4% paraformaldehyde for 30 minutes, permeabilized with 0.3% Triton X-100 for 20 minutes and blocked with 1% bovine serum albumin (BSA) in PBS for 1 hour. The cells were incubated with primary antibody (1:50) in blocking buffer. After sufficient washing, the cells were incubated with FITC-conjugated goat anti-rabbit antibody to detect the primary antibody. The cells were incubated with DAPI for 10 minutes. The resulting images were taken using a laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
For detection of B4GalT5-His and CD24 on the cell surface, the cells were digested and resuspended in 1% BSA in PBS. The cells were incubated with anti-His antibody (1:50) in 1% BSA. After sufficient washing, the cells were incubated with FITC-conjugated goat anti-rabbit antibody and APC-conjugated anti-CD24 antibody for 1 hour. The resulting images were taken using a laser scanning confocal microscope (Carl Zeiss).
14. Animal studies
MCF-7ADR/NC and B4GalT5-overexpressing MCF-7ADR (MCF-7ADR/oe-B4GalT5) cells were subcutaneously transplanted into 14-15 g, 6-week-old female BALB/c nude mice (Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) at a concentration of 2×106 cells per site. Five mice were used in each group for analysis of B4GalT5- induced tumor initiation. The tumor volumes of these mice were measured every week to assess tumor initiation and growth. These tumors were dissected 9 weeks after transplantation and weighed. Then, these tumors were excised, ground, and lysed in loading buffer on ice for 45 minutes. Protein lysates were boiled for 10 minutes and then subjected to western blotting.
15. Statistical analysis
Statistical comparisons between two groups were performed by two-tailed Student’s t tests. The means were calculated using at least three biological replicates. Error bars represent the standard error of the mean for three independent experiments. p-values of < 0.05 were considered statistically significant.
Results
1. B4GalT5 is associated with the stemness of breast carcinoma
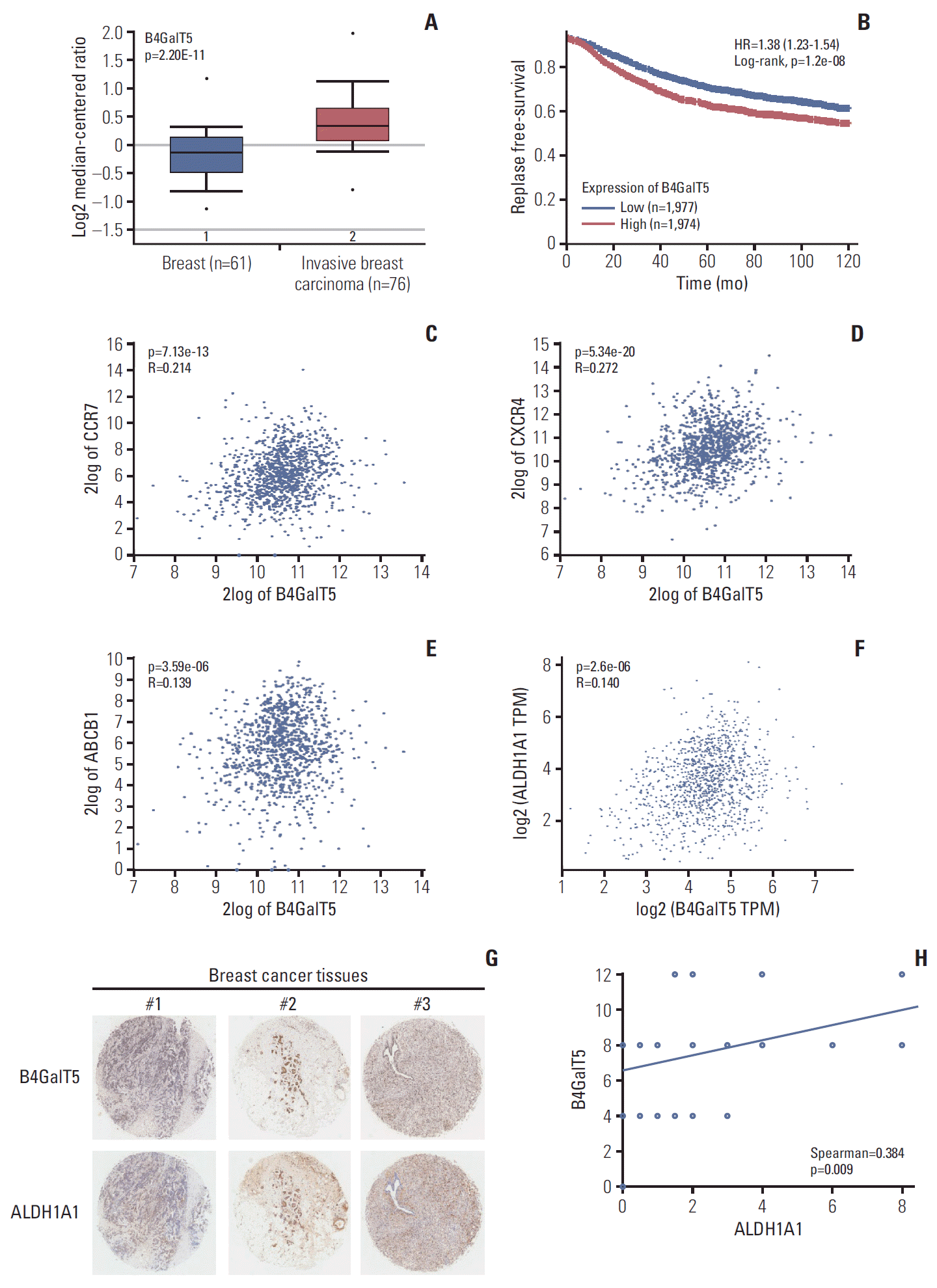
To investigate the relationship between B4GalT5 and BCSCs, we first used online database analysis to compare the B4GalT5 expression level in invasive breast carcinomas with that in normal breast tissues, as CSCs are thought to be closely correlated with the migration and invasiveness of cancer cells [2]. The TCGA breast dataset showed that B4GalT5 was highly expressed in invasive breast carcinomas (Fig. 1A). Kaplan-Meier survival curves for patients with breast cancer (n=3,955) also showed that patients with high expression of B4GalT5 had lower RFS than those with low expression (Fig. 1B, S1 Table). Because BCSCs can establish metastasis and evade traditional anti-tumor therapies [2], we further examined the correlation between B4GalT5 and these malignant phenotypes. Online database analysis showed a positive correlation between B4GalT5 and CC-chemokine receptor 7 (CCR7; R=0.214, p=7.13e-13) (Fig. 1C), CXCR4 (R=0.272, p=5.34e-20) (Fig. 1D), and multidrug resistance protein 1 (ABCB1) (R=0.139, p=3.59e-06) (Fig. 1E), which are involved in tumor metastasis and chemoresistance [10,11]. These results indicate that B4GalT5 is correlated with cancer progression and may be related to BCSCs. ALDH1A1 is related to cancer stem-like features and its high expression and activity has also been proposed as a reliable CSC marker [12]; therefore, we further detected the correlation between B4GalT5 and ALDH1A1. We found that B4GalT5 was positively associated with ALDH1A1 (R=0.14, p=2.6e-06) (Fig. 1F). Since the B4GalT family has seven members, we further investigated the relationship of other B4GalTs and the malignancy of breast carcinoma by online database analysis and found that most B4GalTs (except B4GalT6) were highly expressed in invasive breast carcinomas compared with normal breast tissues (Table 1). Of note, B4GalT5 was the only member positively correlated with the BCSC marker ALDH1A1 (Table 2). For further verification, the expression of B4GalT5 and ALDH1A1 was detected in a tumor microarray containing 45 cancer tissues from patients with invasive breast cancer. Pathologic information regarding these patients is presented in S2 Table. There was a positive correlation between B4GalT5 and ALDH1A1 expression (Spearman=0.384, p=0.009) in 45 patients (Fig. 1G and H). Collectively, these results suggest a critical role of B4GalT5 in regulating the stemness of BCSCs.
2. B4GalT5 is required for maintaining the stemness of breast cancer
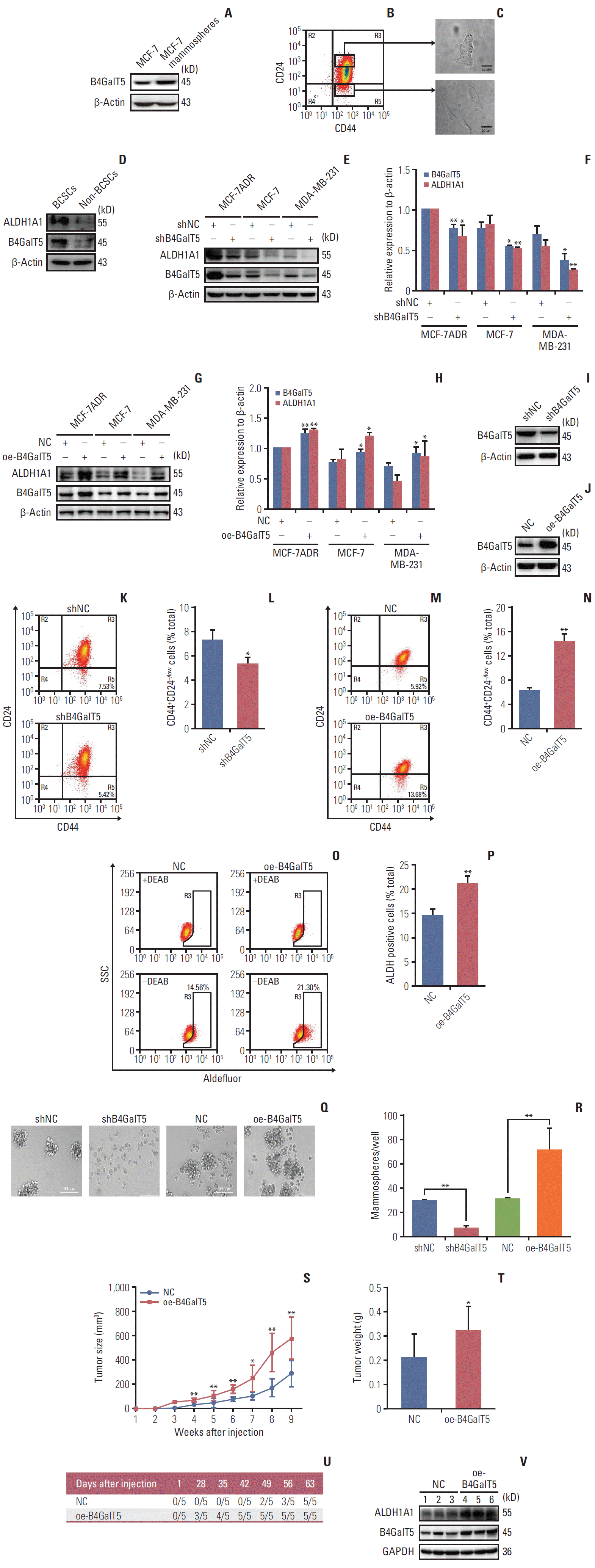
To further investigate the relationship between B4GalT5 and BCSCs, we performed in vitro studies using MCF-7 and MCF-7ADR breast cancer cell lines. CSCs can form mammospheres in serum-free culture medium supplemented with appropriate growth factors due to their capacity for self-renewal and multipotent differentiation; the mammosphere formation assay is also widely used in the isolation and enrichment of CSCs. First, we compared the B4GalT5 expression in MCF-7 mammospheres versus intact MCF-7 cells. As shown in Fig. 2A, western blotting analysis showed that B4GalT5 was highly expressed in the MCF-7 mammospheres.
Because CD44+CD24–/low is identified as the molecular marker of BCSCs [2], we next verified whether the expression of B4GalT5 was associated with CD44+CD24–/low cells. Multiple forms of drug resistance induced by continuous exposure of cells to chemotherapeutics usually generate a high proportion of BCSCs [13]. We therefore chose the adriamycin-resistant breast cancer cell line MCF-7ADR for further research. CD44+CD24–/low cells (BCSCs) and CD44+CD24+ cells (non-BCSCs) were sorted from MCF-7ADR cells aseptically by flow cytometry (Fig. 2B), cultured and subjected to western blotting. Strikingly, the CD44+CD24–/low cells displayed a more malignant phenotype than the CD44+CD24+cells and had a spindle shape and limited cell-cell contacts; in contrast, the CD44+CD24+ cells displayed a cobblestone epithelial morphology (Fig. 2C), which is consistent with a previous report showing that CD44+CD24–/low cells were representative of BCSCs. Furthermore, western blotting analysis showed that B4GalT5 was significantly overexpressed in the CD44+CD24–/low cells versus the CD44+CD24+ cells; in addition, the BCSC marker ALDH1A1 was increased along with overexpression of B4GalT5 (Fig. 2D). Together, these results indicate that B4GalT5 is highly expressed in BCSCs.
To explore the role of B4GalT5 in regulating the stemness of BCSCs, we first detected the effect of B4GalT5 on stem cell marker ALDH1A1 expression in breast cancer cells and found an obvious reduction in ALDH1A1 expression in MCF-7ADR, MCF-7, and MDA-MB-231 cell lines (Fig. 2E and F). In contrast, B4GalT5 overexpression (oe-B4GalT5) significantly augmented ALDH1A1 expression in breast cancer cells (Fig. 2G and H). Moreover, we constructed MCF-7ADR cells with stable knockdown of B4GalT5 (referred to as shB4GalT5, Fig. 2I) and stable overexpression of B4GalT5 (referred to as oe-B4GalT5) (Fig. 2J), followed by in vitro and in vivo studies. We observed a significant decline in the CD44+CD24–/low cells (Fig. 2K and L) in shB4GalT5 cells, as previously observed in siB4GalT5 transfected MCF-7ADR cells. In contrast, B4GalT5 overexpression significantly increased the percentage of CD44+CD24–/low cells (Fig. 2M and N), accompanied by increased ALDH activity (Fig. 2O and P), indicating that B4GalT5 induces the formation of BCSC stemness. Moreover, we performed a mammosphere formation assay to assess the self-renewal ability of BCSCs and the expression level of B4GalT5. We found that the size and number of the mammospheres were markedly restrained in the shB4GalT5 cells, whereas B4GalT5 overexpression promoted the formation of mammospheres (Fig. 2Q and R). It has been reported that CSCs drive tumor initiation, progression, and recurrence [2]. We then subcutaneously injected constructed MCF-7ADR cells into female BALB/c nude mice and found that the group with B4GalT5 overexpression exhibited a dramatic increase in tumor growth and weight compared with the control group (Fig. 2S and T). Specifically, the oe-B4GalT5 cells formed tumors with a volume of 100 mm3 volume earlier than the cells without B4GalT5 overexpression, as plotted in Fig. 2U, further highlighting the strong tumorigenicity conferred by B4GalT5. Furthermore, we found that tumor tissue with B4GalT5 overexpression had higher expression levels of ALDH1A1, as assessed by western blot, than the tissues of the control group (Fig. 2V). Consequently, these results confirm that B4GalT5 plays an important role in maintaining the stemness of BCSCs.
3. B4GalT5 is involved in the inhibition of BCSCs by triptolide
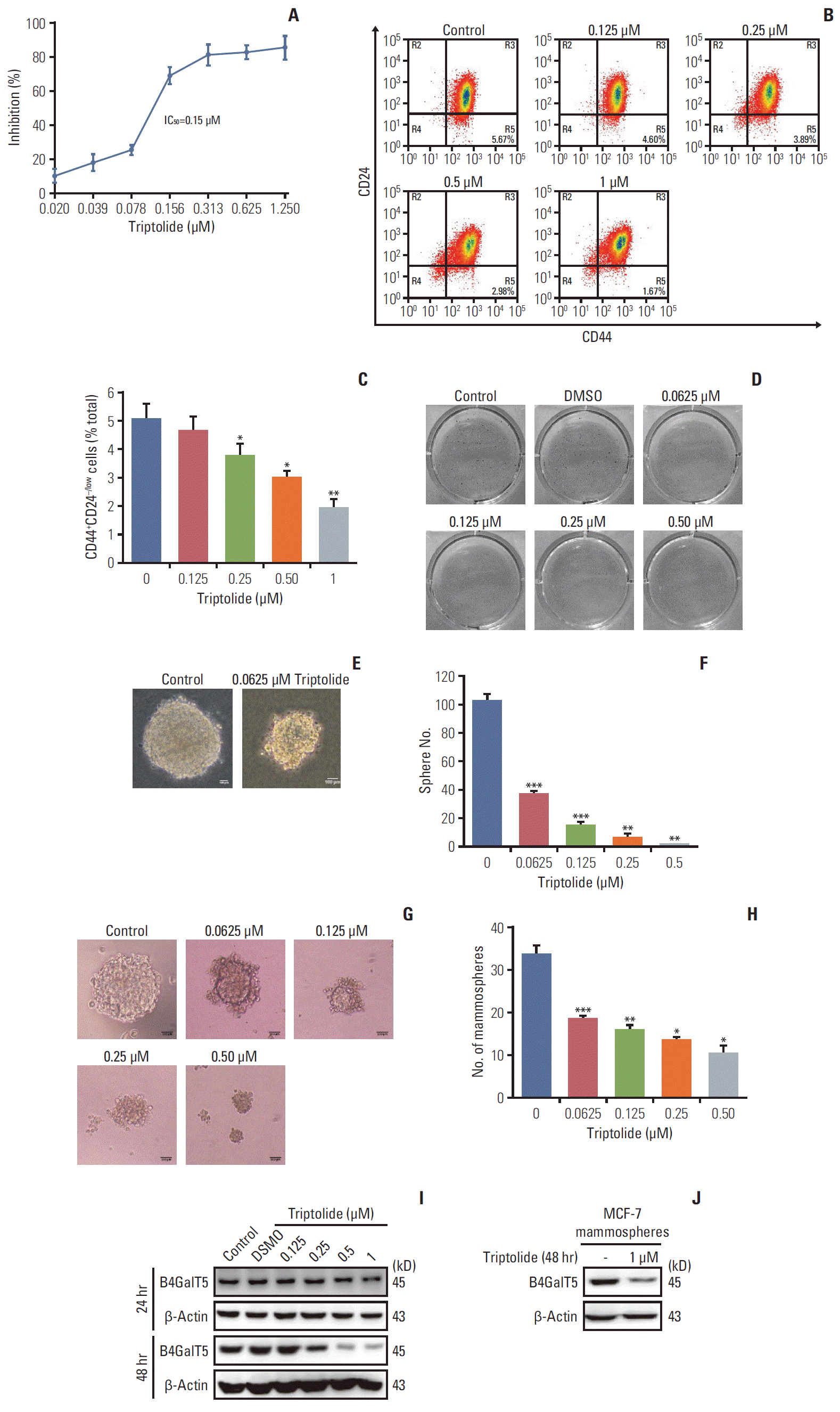
Triptolide, a diterpene triepoxide of extracts derived from the medicinal plant Tripterygium Wilfordii Hook F, has been reported to inhibit the stemness of various cancers, including triple-negative breast cancer and pancreatic cancer [14]. Therefore, we wondered whether B4GalT5 was involved in the inhibition of BCSCs by triptolide. We found that MCF-7ADR cell viability was inhibited by treatment with triptolide, with an IC50 of 0.15 μM (Fig. 3A). In addition, triptolide dramatically reduced the percentage of CD44+CD24–/low cells, suggesting that triptolide significantly eliminated BCSCs (Fig. 3B and C). Furthermore, we found that triptolide apparently suppressed colony growth in soft agar (Fig. 3D-F) and the formation of mammospheres in serum-free medium supplemented with certain cytokines (Fig. 3G and H). All these results confirmed that triptolide was indeed a pharmacological inhibitor of BCSCs. We further investigated whether inhibition of BCSCs by triptolide was accompanied by a decrease in B4GalT5. In the presence of triptolide, the expression level of B4GalT5 was significantly decreased in a dose- and time-dependent manner (Fig. 3I). In particular, B4GalT5 expression in MCF-7 mammospheres was inhibited by treatment with 1 μM triptolide (Fig. 3J). Together, these results further indicate that B4GalT5 is closely correlated with the stemness of breast cancer.
4. Cell surface B4GalT5 is not responsible for the stemness of breast cancer
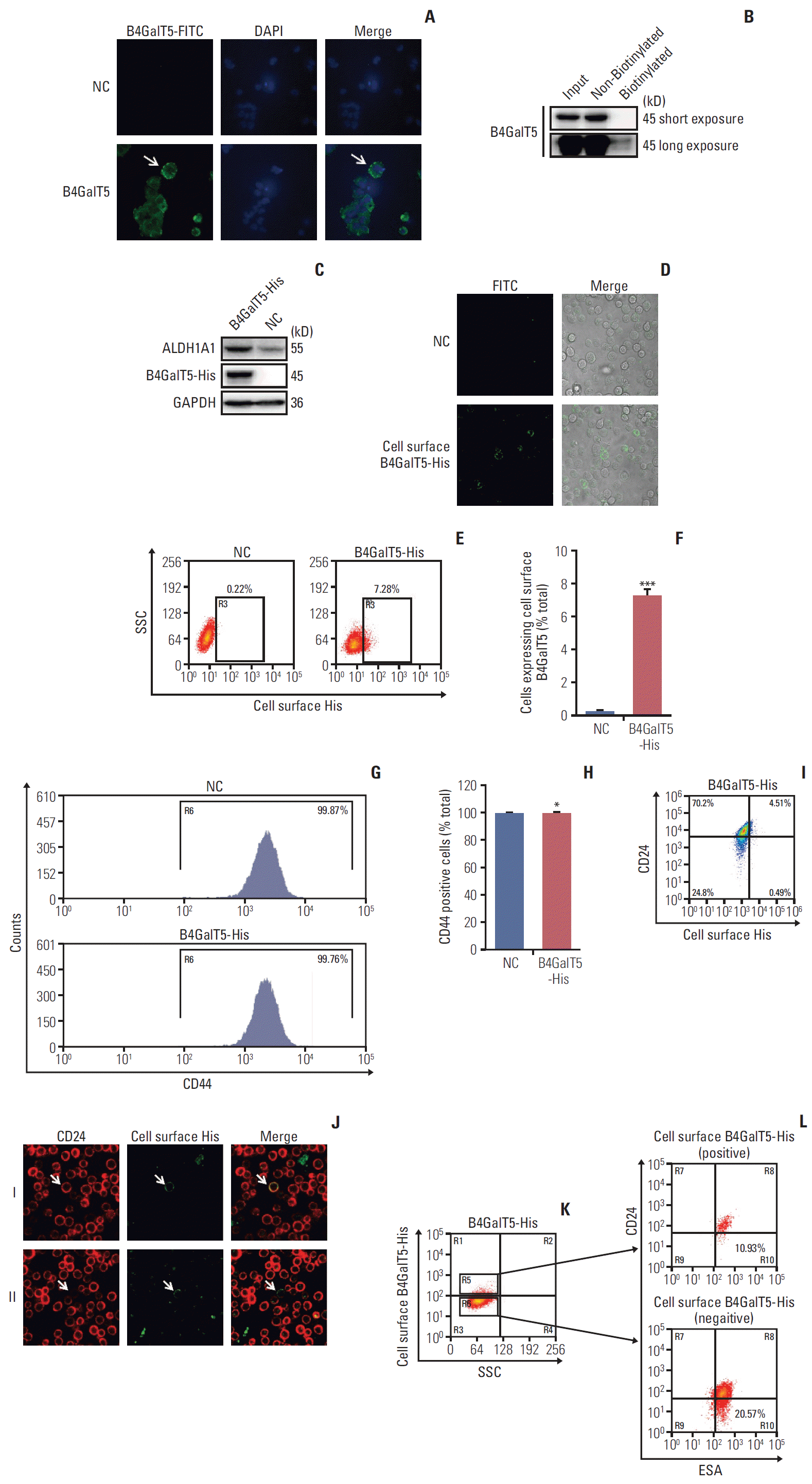
It has been reported that B4GalT show cell surface expression for cell spreading and migration in migratory 3T3 cells [15], indicating that cell surface B4GalTs correlates with cancer malignancy. Thus, we tested whether cell surface B4GalT5 was correlated with the stemness of BCSCs. As shown in Fig. 4A, cell surface B4GalT5 was found in a small portion of MCF-7ADR cells by confocal microscopy. Further, we carried out a cell surface biotinylation assay and proved that the expression level of cell surface B4GalT5 was much lower than that of cytoplasmic B4GalT5 in MCF-7ADR cells (Fig. 4B), indicating that a small proportion of the cells expresses cell surface B4GalT5. These results were consistent with a feature of CSCs, namely a small subset of poorly differentiated tumor cells within a tumor. To further validate the relationship of B4GalT5 expression on the cell surface and BCSCs, we first constructed MCF-7ADR cells that stably overexpressed B4GalT5 with a C-terminal 6×His tag for immunofluorescence staining due to lack of the antibody recognizing its extracellular fragment (Fig. 4C). Western blotting analysis showed that ALDH1A1 was highly expressed in the B4GalT5-His–transfected cells, which suggested the successful construction of cells with active expression of B4GalT5-His (Fig. 4C). We then performed live cell staining using antiHis antibody. B4GalT5-His was detected in a small subset of cells by confocal microscopy without cell membrane permeabilization (Fig. 4D). Furthermore, fluorescence-activated cell sorting (FACS) analysis showed that cell surface B4GalT5-His was expressed in approximately 7.62% of MCF-7ADR cells (Fig. 4E and F). From these results, B4GalT5 was shown to partially localize to the cell membrane with its C-terminus extracellular region, and cell surface B4GalT5 still belonged to type II membrane-bound proteins, consistent with the normal localization of B4GalT5 on the Golgi apparatus [6].
Next, we explored the association between cell surface B4GalT5 and BCSCs by a series of in vitro assays. For convenient detection, we first detected the effect of B4GalT5 on CD44 expression by FACS analysis and observed that CD44 expression in MCF-7ADR cells was almost completely positive and not influenced by B4GalT5 overexpression (Fig. 4G and H). We then investigated the expression of CD24 and B4GalT5 on the cell surface of B4GalT5-His–expressing cells and found that the population with low and negative expression of CD24 (CD44+CD24–/low, BCSCs) accounted for 9.80% of the B4GalT5-His+ cells, whereas this population accounted for 26.10% of the B4GalT5-His–cells (Fig. 4I). We also found that B4GalT5-His+ cells overlapped with not only CD24+ cells but also CD24–/low cells, as observed by confocal microscopy analysis (Fig. 4J). These results suggested that cell surface B4GalT5 may not be a marker of BCSCs. ESA+CD44+CD24–/low cells have also been reported to possess BCSC properties based on their high spheroid formation and in vivo tumor formation ability; hence, the molecular marker group is a more precise way to recognize BCSCs [2]. For further confirmation of the correlation of cell surface B4GalT5 and the stemness of BCSCs, the population rates of ESA+CD44+CD24–/low cells were detected in CD44+ cells positive or negative for B4GalT5-His. As shown in Fig. 4K and L, FACS analysis demonstrated that the population with ESA+CD44+CD24–/low accounted for 10.93% of the B4GalT5-His+ cells, while this population accounted for 20.57% of the B4GalT5-His–cells, which was consistent with the results in Fig. 4H. Together, these results strongly suggest that there is no internal connection between cell surface B4GalT5 and BCSCs, and cell surface B4GalT5 is not responsible for the stemness of breast cancer.
5. B4GalT5 promotes Wnt/β-catenin signaling that is hyperactivated in BCSCs
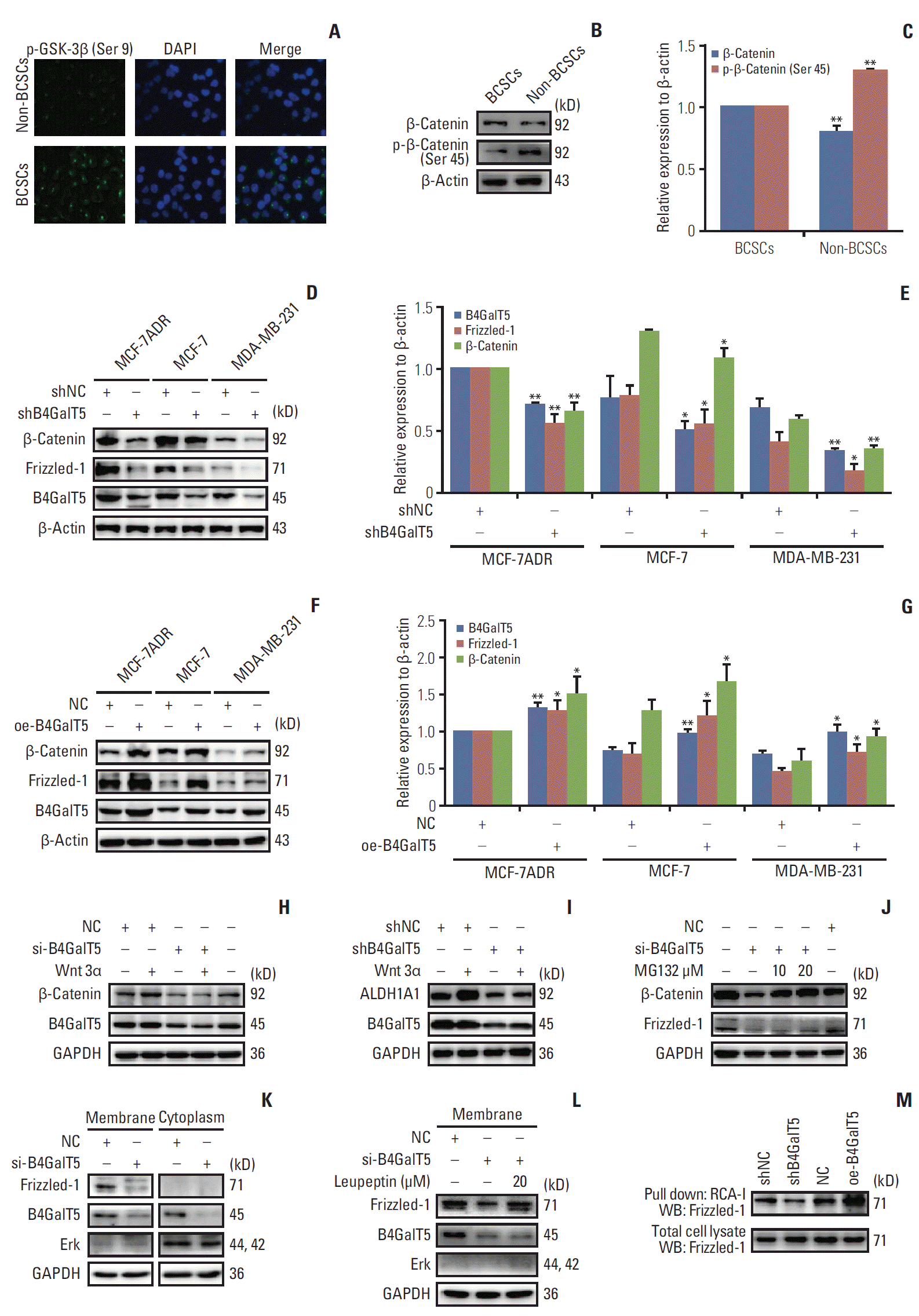
Aberrant Wnt/β-catenin signaling is involved in multiple cancers and closely correlates with cell proliferation, invasion, metastasis, and CSC survival [16]. We tested whether Wnt/β-catenin signaling mediated the regulation of B4GalT5 on the stemness of BCSCs. To validate the relationship between Wnt/β-catenin signaling and BCSCs, we isolated CD44+ CD24–/low cells (BCSCs) and CD44+CD24–/low cells (non-BCSCs) from MCF-7ADR cells using FACS, followed by immunofluorescence and western blotting analysis. GSK3β is reported to be phosphorylated at Ser 9 and thereby inactivated by AKT, resulting in a decrease in β-catenin degradation and activation of Wnt/β-catenin signaling [17]. We found that inactive phosphorylated-Gsk3β (Ser 9) is highly expressed in BCSCs (Fig. 5A). As the substrate of GSK3β, β-catenin showed low phosphorylation at Ser 45 and its expression was upregulated in BCSCs versus non-BCSCs, indicating that the stability of β-catenin was maintained in BCSCs (Fig. 5B and C). All these results suggest that Wnt/β-catenin signaling is hyperactivated in BCSCs compared with non-BCSCs.
To investigate the role of B4GalT5 in regulating Wnt/β-catenin signaling, we first depleted B4GalT5 using B4GalT5 siRNA in multiple breast cancer cell lines, including MCF-7ADR, MCF-7 and MDA-MB-231, and found that the expression levels of the Wnt/β-catenin signaling-related molecules Frizzled-1 and β-catenin were decreased significantly (Fig. 5D and E). In contrast, B4GalT5 overexpression significantly augmented the amounts of Frizzled-1 and β-catenin (Fig. 5F and G). As a canonical Wnt protein, wnt 3α is widely expressed and a representative ligand that interacts with the receptor Frizzled-1 and thereby activates the β-catenin–dependent pathway in Wnt signaling [18]. As shown in Fig. 5H and I, wnt 3α did not successfully induce activation of Wnt/β-catenin signaling and failed to reverse the decrease in stem cell marker ALDH1A1 expression in the B4GalT5-knockdown MCF-7ADR cells, further indicating that B4GalT5 regulates the stemness of breast cancer via activation of Wnt/β-catenin signaling.
We next elucidated the mechanisms underlying the decrease in β-catenin and Frizzled-1 induced by B4GalT5. While Wnt/β-catenin signaling is inactivated, the β-catenin destruction complex is formed and induces β-catenin phosphorylation and degradation by the proteasome system [18]. We observed that the proteasome inhibitor MG132 reversed the decrease in β-catenin in the B4GalT5-knockdown MCF-7ADR cells, whereas the amount of Frizzled-1 was not affected (Fig. 5J). In addition to the proteasomal pathway, the lysosomal system is the other major pathway for protein degradation, especially for cell surface receptors [19]. As the receptor of Wnt signaling, Frizzled-1 was found to be almost completely located on the cell membrane (Fig. 5K), and treatment with the lysosome inhibitor leupeptin clearly blocked B4GalT5 knockdown-induced downregulation of Frizzled-1 in MCF-7ADR cells (Fig. 5L). Together, these results indicate that B4GalT5 knockdown induces degradation of Frizzled-1 via the lysosomal pathway and thereby contributes to β-catenin degradation by the proteasomal pathway, which leads to inactivation of Wnt/β-catenin signaling.
Frizzled-1 is a receptor glycoprotein with N-glycosylation modification, as reported on the Uniprot website (http://www.uniprot.org). Glycans, which are bulky hydrophilic polymers, often contribute to increased stability against proteolysis. Moreover, the covalent binding of glycans to the protein surface may inherently enhance the thermal and kinetic stability of proteins [20]. B4GalT5 participates in the biosynthesis of N-linked oligosaccharides containing galactose residues [6]. Therefore, we assessed whether B4GalT5 participated in the N-glycosylation modification of Frizzled-1 and consequently affected Wnt/β-catenin signaling. RCA-I binds to galactose or N-acetylgalactosamine residues of membrane glycoconjugates, and RCA-I pull-down assays indicated that B4GalT5 knockdown significantly blocked the biosynthesis of galactosyl-oligosaccharides in the glycans of Frizzled-1, while B4GalT5 overexpression increased its biosynthesis (Fig. 5M), indicating a positive correlation between B4GalT5 and Frizzled-1 N-glycosylation. Above all, these findings strongly indicate that B4GalT5 promotes N-glycosylation of Frizzled-1 and its downstream signaling, thereby regulating the stemness of BCSCs.
Discussion
Increasing evidence suggests that BCSCs are involved in tumor progression, recurrence and resistance to chemo- and radiotherapies [3]; thus, identifying specific factors that can regulate BCSC properties is important for reducing recurrence and thereby improving the therapeutic effect on breast cancer. In this paper, we demonstrated that B4GalT5-mediated regulation of BCSCs positively correlated with malignant phenotypes and poor prognosis of breast cancer by a series of online database analyses and IHC analyses followed by in vitro and vivo studies.
A characteristic feature of the malignant transformation of tumor cells is the alteration of highly branched N-linked oligosaccharides on glycoproteins [4]. Abnormal expression of B4Gal-T1-T7 has been reported to be involved in changes in highly branched N-linked oligosaccharides, which are implicated in a number of diseases, including various cancers, inflammation, and viral infection [6]. For example, B4GalT2 is a critical effector involved in neuronal development, congenital muscular dystrophies and astrocytoma [21]. B4GalT4 overexpression is closely associated with colorectal cancer metastasis and poor prognosis in patients [22]. B4GalT5 and B4GalT6 are responsible for the production of lactosylceramide (LacCer) synthase, which functions in the initial step of ganglioside biosynthesis [23]. Moreover, B4GalT5 participates in the synthesis of both N-linked oligosaccharides and various glycolipids, contributing to highly galactosylated cell surface proteins that are associated with extraembryonic development and the tumorigenesis of astrocytoma, melanoma and glioma [6]. Finally, B4GalT7 regulates the synthesis of glycosaminoglycans that play a central role in many pathophysiological events [24]. In this paper, we found that the expression levels of B4GalTs (except B4GalT6) were upregulated in invasive breast carcinomas compared with mammary tissues, yet only B4GalT5 was found to be positively associated with the BCSC marker ALDH1A1, indicating that B4GalT5 displays CSC-related specificity and might play important roles in breast cancer.
Several studies have reported that Wnt/β-catenin signaling is aberrantly activated in various human malignancies, including breast, colorectal, gastric, lung, ovary, pancreatic, prostate and uterine cancers, leukemia and melanoma, and is involved in CSC survival, bulk tumor expansion and metastasis [25]. In this pathway, the Wnt ligand interacts with the receptor Frizzled-1, inhibiting the degradation of β-catenin and subsequently translocating into the cell nucleus, which can regulate the transcription of a number of target genes related to cancer development [25]. Our studies also confirmed that Wnt/β-catenin signaling was more active in BCSCs than in non-BCSCs. As the receptor of Wnt/β-catenin signaling, Frizzled-1 shows N-glycosylation that has been demonstrated to play an important role in regulating biological activity, intracellular targeting, protein folding, and maintenance of protein stability [26]. We propose for the first time that B4GalT5 mediates the biosynthesis of galactosyloligosaccharides in the N-glycosylation modification of Frizzled-1, which contributes to the activation of Wnt/β-catenin signaling and thereby regulates the stemness of breast cancer. Furthermore, we found that B4GalT4 knockdown promoted Frizzled-1 degradation by the lysosomal pathway and then contributed to the degradation of β-catenin through the proteasomal pathway. Previous studies have reported that incomplete N-glycosylation affects the stability of cell surface proteins, such as N-cadherin and programmed death-ligand 1, and leads to their ubiquitylation and subsequent proteasomal degradation [27,28], which is inconsistent with our findings. Since proteins on the cell membrane recycle from endosomes to the plasma membrane or are transported to lysosomes for degradation [29], we believe that incomplete N-glycosylation resulting from the depletion of B4GalT5 affects the stability of Frizzled-1 located in the cell membrane and eventually results in its degradation through the lysosomal pathway.
In addition to the location of B4GalTs in the Golgi complex, glycosyltransferases are present on the cell surface, where they function as adhesion molecules that are involved in cell-to-cell and cell-to-extracellular matrix interactions as well as cell spreading and migration, and associated with signal transduction cascades [15,30]. Consistent with these previous studies, our data also showed that B4GalT5 was located on the cell surface of MCF-7ADR cells and first confirmed that it is a type II membrane-bound protein. As B4GalT5, located partly on the cell surface, was related to cell spreading and migration [15], cell surface B4GalT5 may be a molecular marker for the stemness of BCSCs. However, flow cytometry and immunofluorescence assays revealed that cell surface B4GalT5 was not responsible for the stemness of BCSC. These results in turn demonstrated that B4GalT5 canonically located in the Golgi complex, which affected the level of Frizzled-1 by glycosylation modification on the cell surface, was responsible for the stemness of breast cancer through Wnt/β-catenin signaling. The biological function of cell surface B4GalT5 may only mediate spreading and migration of a small number of cells. The finding of a correlation between glycosyltransferase distribution and its function in BCSCs provides more foundations for drug design to target BCBCs.
In conclusion, we showed that B4GalT5 is overexpressed in invasive breast carcinomas, positively associated with poor survival, and closely related to the stemness of BCSCs. We have demonstrated for the first time that B4GalT5 regulates BCSC properties by affecting the N-glycosylation modification of Frizzled-1, thereby affecting its stability on the cell membrane and inhibiting downstream signaling in BCSCs, which is independent of the B4GalT5 location in cells. Our findings provide novel insights into the important role of B4GalT5 in BCSCs. This study also suggests that targeting B4GalT5 may be a promising strategy to suppress or eliminate CSCs in breast cancer.
 XML Download
XML Download