

 PDF
PDF Citation
Citation Print
Print


Introduction
Since the role of the DNA damage response (DDR) pathway in cancer survival was discovered, targeting the DDR pathway has been evaluated as a treatment strategy for diverse cancers [1]. As the core members of the DDR signaling network, poly(ADP-ribose) polymerase (PARP), ataxia telangiectasia mutated (ATM), ataxia telangiectasia mutated and Rad3 related (ATR), and the nuclear kinase WEE1 control DNA damage repair via regulating cell cycle progression, apoptosis, chromatin remodeling, and oncogenic signaling pathways [1]. In terms of the crucial actions of these DDR-related mediators, PARP/ATM/ATR/WEE1 inhibitors are under clinical development for solid tumors. Although targeting the DDR pathway is an area of intense research focus, little research has been devoted to its role in biliary tract cancer (BTC), an aggressive cancer.
To date, the first-line treatment for patients with advanced BTC is a gemcitabine/cisplatin combination, of which clinical outcome is limited [2,3]. Given the huge unmet medical needs, potential targeted agents should be thoroughly developed in BTC. Previous studies reported that TP53 and retinoblastoma protein (RB) module alterations were found in 33.9% and 11.7% of patients with BTC, respectively [4]. One of the major functions of TP53 and RB is control of the G1-S cell cycle transition [5]. However, because of G1/S checkpoint dysfunction, the cells were more dependent on G2/M checkpoint proteins, such as WEE1, for survival [6,7]. In addition, alterations in DNA damage repair-related genes, including breast cancer 1/2 (BRCA1/2), BRCA1 associated protein-1, and histone-lysine N-methyltransferase 2D, were also found in approximately 20% of patients with BTC [4]. These characteristics support BTC as a good model for the development of DDR-targeted agents, especially WEE1 inhibitors.
The nuclear kinase WEE1 is an important cell cycle regulator that inhibits cyclin-dependent kinase 1 activity, and it participates in the homologous recombinant pathway to repair DNA damage [1,6]. Because of this role, preclinical studies have tested WEE1 inhibition in DDR-deficient cancer cells, which showed that WEE1 blockade could induce strong anti-tumor effects through enhancing DNA damage, G2/M arrest, and apoptosis [8,9]. However, WEE1 inhibition has not yet been evaluated in BTC cells.
In our work, the anti-tumor effects of AZD1775 [6], a potent WEE1 kinase inhibitor, in BTC were explored. In the study, we aimed to elucidate the mechanism of sensitivity to WEE1 inhibitor in BTC using in vitro and in vivo experiments.
Materials and Methods
1. Human cell lines and reagents
Nine human BTC cell lines were utilized in this study. SNU245, SNU308, SNU478, SNU869, and SNU1196 cells were purchased from Korean Cell Line Bank (Seoul, Korea). HuCCT-1 and TFK-1 cells were obtained from RIKEN BioResource Center (Ibaraki, Japan). The patient-derived cell lines SNU2670 and SNU2773 were successfully established as described previously [10]. All cells were cultured in RPMI1640 medium (Welgen Inc., Gyeongsan, Korea) containing 10% fetal bovine serum and 10 μg/mL gentamicin at 37°C under 5% CO2. WEE1 (AZD1775), ATR (AZD6738), and ATM (AZD0156) inhibitors were kindly provided by AstraZeneca (Macclesfield, Cheshire, UK).
2. Cell viability assay
Cells were seeded in 96-well plates and incubated overnight at 37°C. The cells were exposed to increasing concentrations of AZD1775 alone or in combination with AZD6738 (ATR inhibitor) or AZD0156 (ATM inhibitor) for 3 days. Next, 50 μL of 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) solution (Sigma-Aldrich, St. Louis, MO) were added to each well, and plates were incubated at 37°C for 4 hours. The medium was removed, and 150 μL of dimethyl sulfoxide were added to each well. Cell viability was measured at 540 nm using a VersaMax Microplate Reader (Molecular Devices, Sunnyvale, CA). The experiments were performed in triplicate.
3. Colony-forming assay
Cells were seeded in 6-well plates and exposed to various concentrations of AZD1775. After 10 days, the colonies were stained with Coomassie blue for 2 hours and counted using Gel Doc system software (Bio-Rad, Hercules, CA). Each experiment was repeated three times.
4. Western blot analysis
Cells were seeded in 60-mm dishes and treated with AZD1775, AZD6738, or both for 24 hours. The cells were harvested and lysed in RIPA buffer containing protease inhibitors on ice for 30 minutes. The proteins were extracted, and equal amounts of proteins were used for western blot analyses. Primary antibodies against the following molecules were purchased from Cell Signaling Technology (Beverley, MA): WEE1 (#4936), p-WEE1-Ser642 (#4910), ATR (#2790), phosphorylated ATR-Ser428 (#2853), Chk1 (#2360), phosphorylated Chk1-Ser345 (#2341), PARP (#9532), caspase-7 (#9492), phosphorylated AKT-Ser473 (#9271), AKT (#9272), phosphorylated CDC25C-Ser216 (#9528); CDC25C (#4688); phosphorylated CDC2 (#9111), CDC2 (#9112), and p21 (#2947). β-Actin antibody was purchased from Sigma-Aldrich. Anti-ATM (#ab78) and phosphorylated ATM-Ser1981 (#ab81292) antibodies were obtained from Abcam Bioscience (Cambridge, UK). Anti-γH2AX antibody (#05-636) was bought from Millipore (Billerica, MA). Secondary antibodies were acquired from Thermo Fisher Scientific Inc. (Waltham, MA).
5. Cell cycle analysis
Cells were seeded in 60-mm dishes and treated with various concentrations of AZD1775 for 24 hours. The cells were harvested and fixed with 70% ethanol at −20°C. After 2 days, 7 μL of RNase A (20 mg/mL, Invitrogen, Carlsbad, CA) were added to each well and incubated for 10 minutes at 37°C. The cells were stained with 13 μL of propidium iodide (SigmaAldrich) and analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Each experiment was repeated three times.
6. Phospho-histone H3 staining assay
Cells were seeded in 60-mm dishes and incubated with AZD1775 for 24 hours. Then, cells were fixed with 70% ethanol at least 4 hours at −20°C. After washing with staining buffer (#420201, BioLegend, San Diego, CA), 20 μL of phospho-histone H3 (p-HH3) antibody (#558217, BD Bioscience) and 80 μL of staining buffer were added for 20 minutes at room temperature. Cells were washed with staining buffer once and incubated with 300 μL of staining buffer and 3.5 μL of RNase A for 10 minutes at 37°C. Next, 13 μL of propidium iodide were added, and cells were analyzed using a FACSCalibur flow cytometer. Each experiment was repeated three times.
7. Transwell migration assay
Migration assays were conducted using 6.5-mm Transwell insert chambers with 8-μm-pore polycarbonate membranes (#CLS3422, Sigma-Aldrich). Cells were seeded into the upper chamber in 200 μL of medium containing 0.1% fetal bovine serum (FBS), and 500 μL of medium containing 10% FBS were added to the lower chamber. AZD1775 was added to the upper chamber, and the plates were incubated at 37°C for 24 hours. The non-migrated cells remaining on the upper side of the membranes were removed with cotton swabs, and the filters were fixed with 4% paraformaldehyde (#P2031, Biosesang, Seongnam, Korea) for 20 minutes at room temperature. The membranes were then incubated in 1% crystal violet solution (#V5265, Sigma-Aldrich) for 10 minutes. The cells were visualized using a microscope and photographed, after which they were then dissolved by incubating the membranes in 300 μL of 33% acetic acid (#1.00063.2511, Merck, Darmstadt, Germany) for 10 minutes. The liquid was collected, and the absorbance at 573 nm was measured using a microplate reader. The experiment was repeated three times.
8. Alkaline comet assay
Cells were exposed to 1 μmol/L AZD1775, 1 μmol/L AZD6738, or both for 24 hours. Then, the cells were harvested, and alkaline comet assay was performed as previously described [11]. The tail moment and intensity were measured using the Comet Assay IV program (Andor Technology, Belfast, UK). Three independent experiments were analyzed for each condition.
9. In vivo experiments
Animal experiments were performed at the Institute for Experimental Animals, College of Medicine, Seoul National University (Seoul, Korea) according to institutional guidelines, with prior approval from the Institutional Animal Care and Use Committee. Four-week-old female athymic nude mice were purchased from Orient Bio Inc. (Seongnam, Korea). HuCCT-1 and SNU2670 xenograft model mice were established via subcutaneous inoculation of 1×107 cells in 100 μL of phosphate buffered saline. When the tumor volume reached 200 mm3, the mice were randomly divided into two or four groups of five mice. AZD1775 (25, 60, or 90 mg/kg) and AZD6738 (25 mg/kg) were administered orally once a day for 3 weeks (5 days on/2 days off), and the control group was treated with vehicle (2-hydroxypropyl-β-cyclodextrin solution, Sigma-Aldrich) via oral gavage. Body weights and tumor sizes were measured every other day. The tumor volume was calculated using the following formula: tumor volume=[(width)2×height]/2.
10. Immunohistochemistry
Sections of paraffin-embedded xenograft tumor tissues were deparaffinized and dehydrated. Immunohistochemical (IHC) detection of p-ATR (#2853, Cell Signaling) expression and proliferating cells was conducted using an anti-Ki-67 antibody (GeneTex Inc., Irvine, CA) at a dilution of 1:100. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assays were conducted for the immunohistochemical detection of apoptosis using an ApopTag In Situ Apoptosis Detection Kit (EMD Millipore) according to the manufacturer’s protocol.
11. Statistical analysis
Statistical analyses were conducted using SigmaPlot ver. 10.0 (Systat Software Inc., San Jose, CA). Experimental data are presented as the mean±standard error. All statistical tests were two-sided. Differences were considered statistically significant at p < 0.05. The half-maximal inhibitory concentration (IC50) of agents was also analyzed using SigmaPlot software. Combined drug effects were analyzed by calculating the combination index (CI) using CalcuSyn software (Biosoft, Cambridge, UK). CIs of < 1, 1, and > 1 indicate synergistic, additive, and antagonistic effects, respectively.
12. Ethical statement
Animal experiments were performed at the Biomedical Center for Animal Resource Development of Seoul National University (Seoul, Korea) according to institutional guidelines, and prior approval of the study protocol was obtained from the Institutional Animal Care and Use Committee.
Results
1. The anti-proliferative effects of WEE1 inhibition in BTC cells
To evaluate whether WEE1 inhibition has anti-proliferative effects in BTC cells, the MTT assay was performed after treating cells with various concentrations of AZD1775 for 72 hours. Among the nine cell lines, viability was more strongly suppressed by AZD1775 in SNU308, SNU478, SNU869, SNU1196, HuCCT-1, and TFK-1 cells than in SNU245, SNU2670, and SNU2773 cells (Fig. 1A and B). According to the sensitivity, we selected four cell lines for the subsequent experiments. SNU308 (KRAS/TP53 mutation) and HuCCT-1 (KRAS/TP53/RAD51/BRCA2 mutation) are relatively sensitive cells, and SNU2773 (HER2 positive) and SNU2670 (BRAF mutation/ HER2 positive) are relatively insensitive cells.
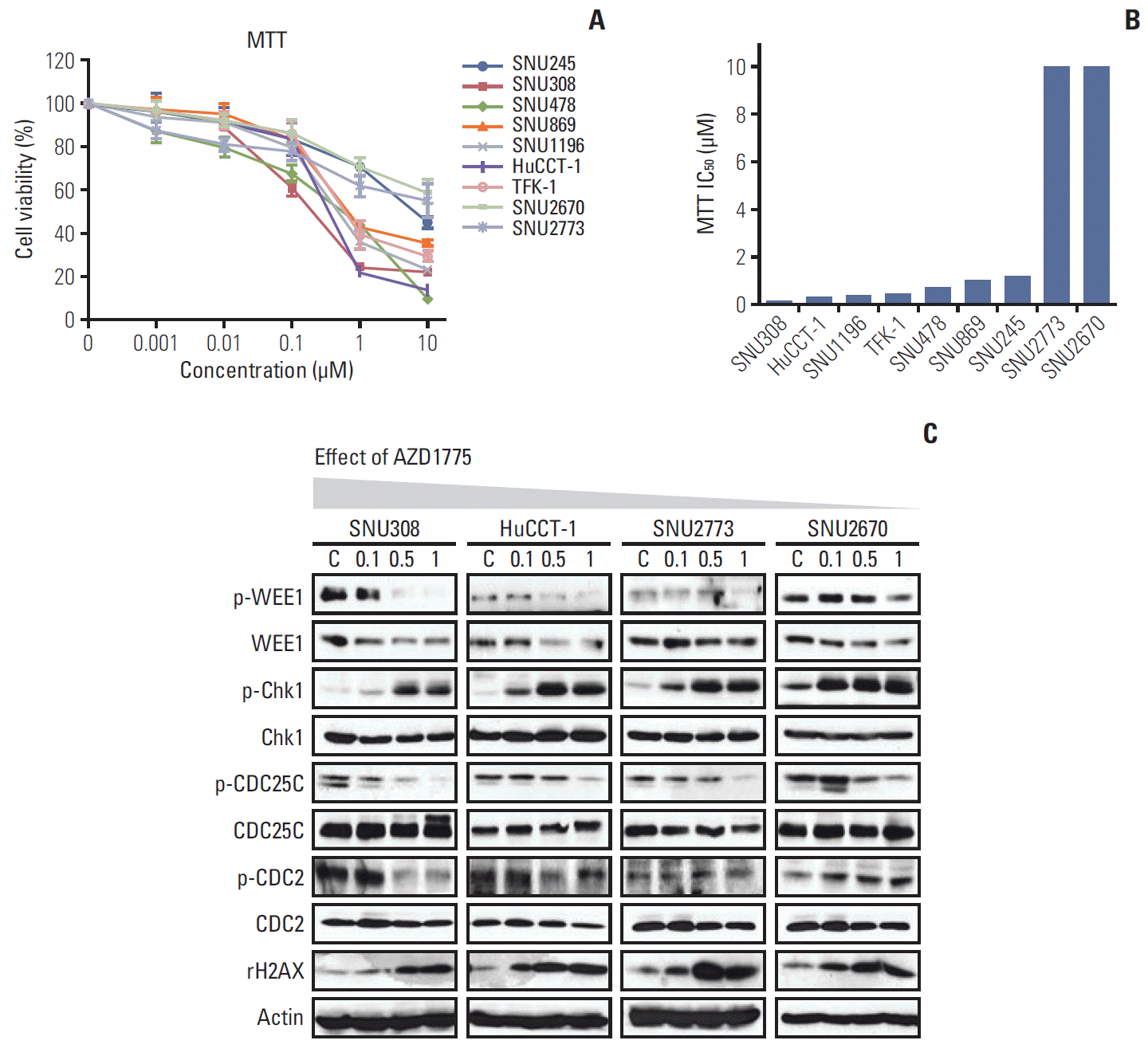
Fig. 1.
Anti-proliferative effect of AZD1775 in biliary tract cancer (BTC) cells. (A) The anti-proliferative effects of AZD1775 in nine BTC cell lines were evaluated using the 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) assay after 72 hours. (B) The half-maximal inhibitory concentration (IC50) of AZD1775 in the MTT assay was calculated. (C) Western blot analyses of the effects of AZD1775 on signaling pathways in four BTC cell lines. Cells were treated with increasing concentrations of AZD1775 (0, 0.1, 0.5, and 1 μM) for 24 hours.
![]()
The colony-forming assay was conducted 10 days after treatment with AZD1775. As shown in S1 Fig., AZD1775 dramatically reduced colony formation in all four cell lines, especially SNU308 cells (S1A and S1B Fig.). To confirm this observation, we detected the related DNA damage signals at 24 hours after treatment (Fig. 1C). Interestingly, AZD1775 as a WEE1 kinase inhibitor downregulated WEE1 total levels in all four cell lines. According to previous research on AZD1775, probably because reduced levels of transcription factors or PLK1 prevented WEE1 expression [12,13]. In addition, increased p-Chk1 and γ-H2AX levels and decreased p-CDC2 level was observed in all four cells. However, p-CDC25C was reduced in all four cell lines. In general, CDC25C phosphorylation at Ser216 is accompanied by an increase in p-Chk1/p-Chk2, but we have not observed a canonical signaling relationship between them in BTC cell lines.
2. WEE1 inhibition induces S phase arrest, apoptosis and suppresses migration in sensitive cells
To analyze AZD1775-mediated cell cycle regulation, cells were incubated with AZD1775 for 24 hours and then examined using flow cytometry. WEE1 inhibition led to significant S phase arrest in SNU308, HuCCT-1, and SNU2773 cells but not in SNU2670 cells (Fig. 2A). But AZD1775 induced S phase arrest in SNU2670 cells within 48 hours, and the sub-G1 population still no changed (data not shown, p < 0.01). The number of p-HH3–positive cells in S phase was remarkably increased for SNU308, HuCCT-1, and SNU2773 cells (Fig. 2B). We also observed increased sub-G1 population, which represents apoptosis, in SNU308 and HuCCT-1 cells (Fig. 2A). In addition, we detected related signals under the same conditions. Compared with the findings in SNU2773 and SNU2670 cells, cleaved PARP and caspase-7 levels, as well as p21 levels, were strongly increased in SNU308 and HuCCT-1 cells (Fig. 2C).
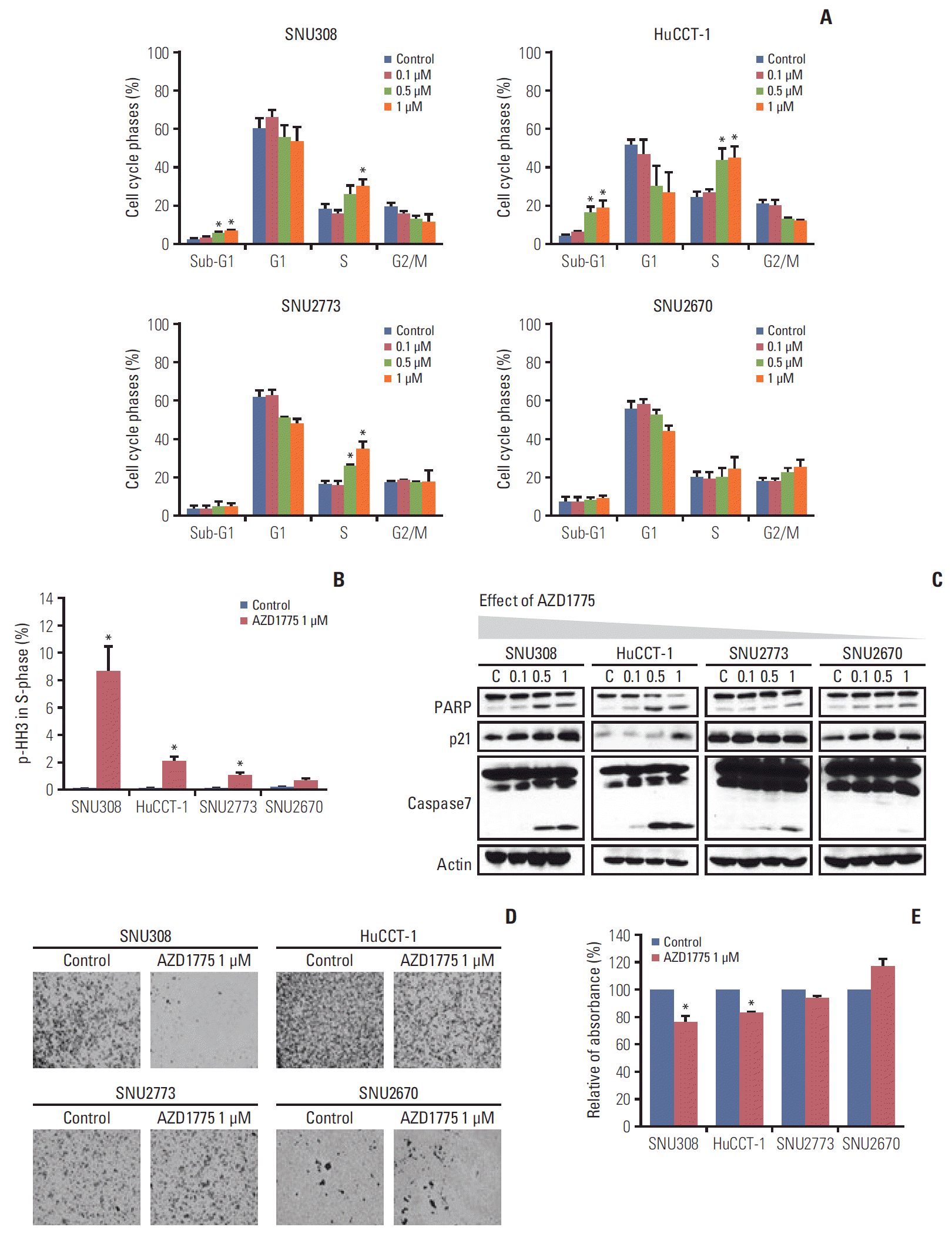
Fig. 2.
The effect of AZD1775 alone in biliary tract cancer (BTC) cells. (A) Cell cycle analyses were performed by flow cytometry after treatment with increasing concentrations of AZD1775 (0, 0.1, 0.5, and 1 μM) for 24 hours. The data represent three independent experiments. *p < 0.05. (B) The phospho-histone H3 (p-HH3)–positive population in S phase was analyzed using flow cytometry after treatment with AZD1775 (0 and 1 μM) for 24 hours. The data represent three independent experiments. *p < 0.05. (C) Western blot analyses of the effects of AZD1775 on cell cycle pathways in four BTC cell lines. Cells were treated with increasing concentrations of AZD1775 (0, 0.1, 0.5, and 1 μM) for 24 hours. PARP, poly(ADP-ribose) polymerase. (D, E) Transwell migration assays were conducted with or without AZD1775 treatment (1 μM). The image was captured at 24 hours after treatment. The percentage of migratory cells was analyzed after images were captured. Experiments were repeated three times. *p < 0.05.
![]()
Next, the Transwell migration assay was performed because the effects of DDR-targeted agents on cell migration have rarely been reported. Interestingly, WEE1 inhibition resulted in significant blockade of cell migration within 24 hours in both SNU308 and HuCCT-1 cells, but not in SNU2773 and SNU2670 cells (Fig. 2D and E).
3. ATM/ATR activation correlates with sensitivity of BTC cells to WEE1 inhibition
Having examined the effects of AZD1775 in BTC cells, we sought to explain the difference in sensitivity of the cells to WEE1 inhibition. We measured ATM and ATR levels after treatment with AZD1775 in the four cell lines. Interestingly, p-ATM and p-ATR expression was concentration-dependently downregulated in SNU308 and HuCCT-1 cells (Fig. 3A). On the contrary, both p-ATM and p-ATR levels were increased in SNU2773 and SNU2670 cells after treatment with AZD1775 (Fig. 3A). In DNA damage repair pathways, the ATM/ATR could be activated by many partners [14,15]. WEE1 inhibition might reduce positive controller such as TIP60, ETAA1, and TopBP1, or enhance negative controller like histone linker 1.2. These results provided us the rationale to assess combination treatment with WEE1 and ATR (AZD6738)/ATM (AZD0156) inhibitors in BTC cells. Next, to evaluate whether the combination regimens were synergistic, we conducted MTT assays for 72 hours. As shown in Fig. 3B, strong synergistic effects were observed in SNU2773 and SNU2670 cells for combined treatment with AZD1775/AZD6738 or AZD1775/AZD0156. However, AZD1775/AZD6738 was slightly synergistic in SNU308 and HuCCT-1 cells.
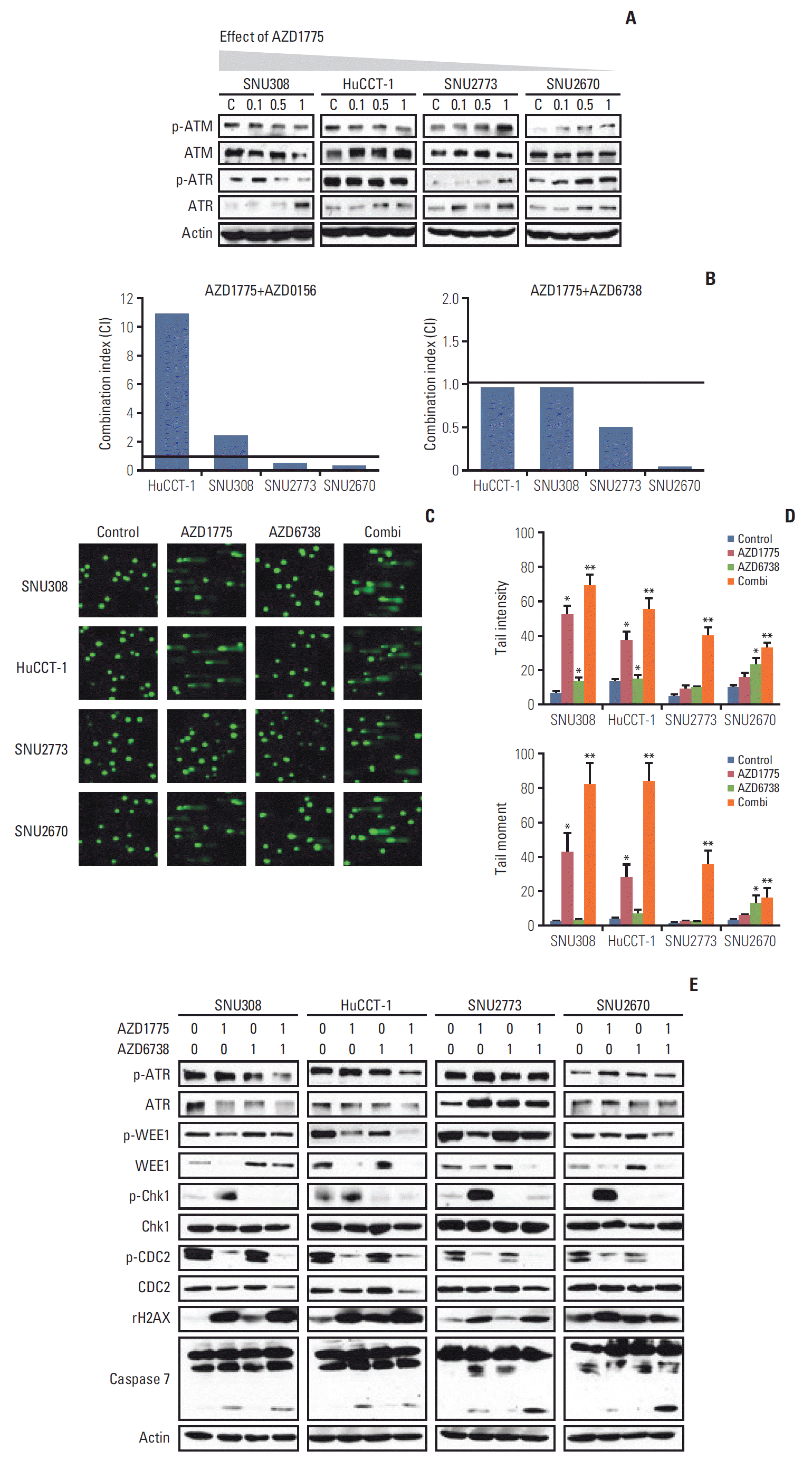
Fig. 3.
Combination effects of WEE1 and ataxia telangiectasia mutated and Rad3 related (ATR) inhibitors in biliary tract cancer. (A) Western blotting was performed after treated with various concentrations of AZD1775 (0, 0.1, 0.5, and 1 μM) for 24 hours. ATM, ataxia telangiectasia mutated. (B) The combination indices (CIs) in the 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide assay after combination treatment with AZD1775 (0, 0.001, 0.01, 0.1, 1, and 10 μM) and AZD0156 (0, 0.001, 0.01, 0.1, 1, and 10 μM)/AZD6738 (0, 0.001, 0.01, 0.1, 1, and 10 μM) for 72 hours. CI value was analyzed using CalcuSyn software. CI > 1, 1, and < 1 indicate antagonistic, additive, and synergistic effects, respectively. (C, D) Comet assays were conducted after treatment with AZD1775 (1 μM), AZD6738 (1 μM), or both for 24 hours. The tail intensity and moment were analyzed using the Comet Assay IV program. Experiments were repeated three times. *p < 0.05, **p < 0.01. (E) Cells were incubated with AZD1775 (1 μM), AZD6738 (1 μM), or both for 24 hours, and then western blotting was performed.
![]()
To confirm the effects of the combination treatments on DNA damage, a comet assay was performed. The tail intensity and moment data revealed that both AZD1775/AZD6738 and AZD1775/AZD0156 synergistically induced DNA damage in all four cell lines (Fig 3C and D, S2 Fig.). In line with the anti-proliferative assay, AZD1775 mono-treatment significantly induced DNA damage in sensitive cells not in less sensitive cells. We speculated that DNA damage might be repaired through ATM or ATR activation in SNU2773 and SNU2670 cells. Given that AZD6738 has more potent antitumor effect in clinical trials compared to AZD0156 [15], we detected related signals under the same conditions using the AZD1775/AZD6738 combination. AZD1775-mediated p-ATR upregulation was reduced by co-treatment with AZD6738. We also found that p-Chk1 was increased by AZD1775 in all four cell lines, but AZD6738 strongly blocked p-Chk1 expression. Co-treatment strongly downregulated p-CDC2 expression and increased γ-H2AX accumulation in all cell lines. Cleaved caspase-7 expression was more increased by combined treatment in less sensitive cells (Fig. 3E). We also conducted Transwell migration assay under the combination treatment. Although combination treatment did not show a synergism, the combination treatment has strong ability to suppress migration in SNU2773 and SNU2670 cell lines (data not shown, p < 0.05).
4. Co-targeting ATR and WEE1 exhibited synergistic antitumor growth effects in xenograft models
After evaluating the combination regimens in vitro, we established two xenograft models using HuCCT-1 and SNU2670 cell lines to confirm our findings in vivo. AZD1775 monotherapy suppressed tumor growth in HuCCT-1 xenograft mice at a low dose (25 mg/kg); however, the therapy did not inhibit tumor growth in SNU2670 xenograft mice (Fig. 4A). At a high dose (90 mg/kg) in relatively sensitive HuCCT-1 xenograft mice, AZD1775 nearly blocked tumor growth. In relatively insensitive SNU2670 xenograft mice, AZD1775 also inhibited tumor growth.
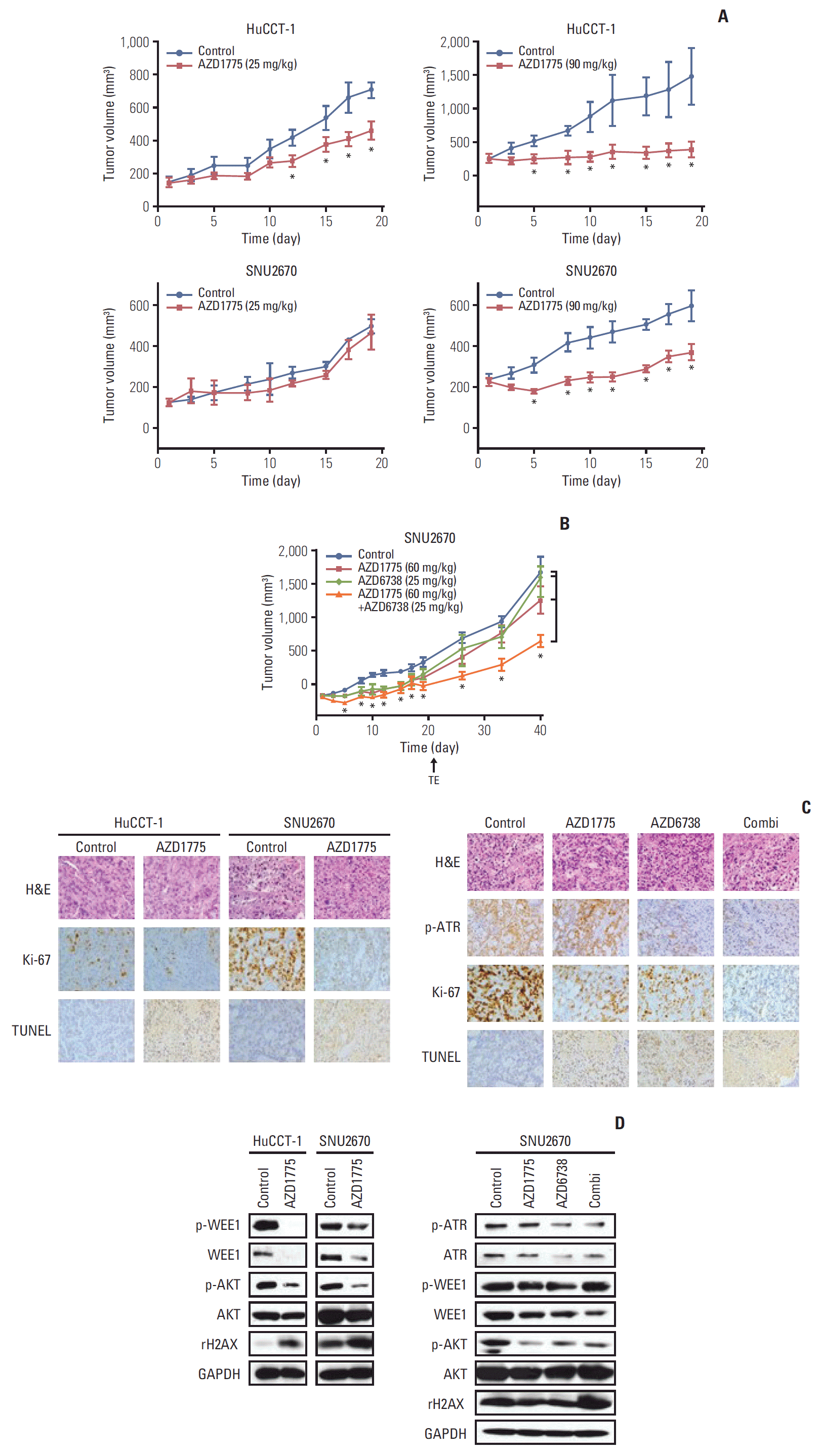
Fig. 4.
Anti-tumor effects of AZD1775 alone and in combination with AZD6738 in xenograft mouse models. (A) HuCCT-1 and SNU2670 xenograft mice were administrated vehicle or AZD1775 (25 or 90 mg/kg) for 3 weeks (5 days on/2 days off). Each group contained five mice. *p < 0.05. (B) SNU2670 xenograft mice were treated with vehicle, AZD1775 (60 mg/kg), AZD6738 (25 mg/kg), or both AZD1775 and AZD6738 for 3 weeks (5 days on/2 days off). Each group contained five mice. TE, end of treatment. *p < 0.05. (C) H&E staining (×400) and immunohistochemical analysis (×400) of p-ataxia telangiectasia mutated and Rad3 related (ATR), Ki-67, and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) expression were performed using isolated HuCCT-1 and SNU2670 tumors. (D) Western blot analysis of various proteins in isolated tumors.
![]()
Based on this observation, we conducted combination treatment using AZD1775 (60 mg/kg) and AZD6738 (25 mg/kg) in SNU2670 xenograft mice. Notably, neither AZD1775 nor AZD6738 monotherapy efficiently suppressed tumor growth, but the combination regimen significantly inhibited tumor growth (Fig. 4B). During the experiment, we did not observe body weight loss in any animals (S3 Fig.).
At the end of the experiment, the tumors of the mice were isolated, and p-ATR, Ki-67, and TUNEL expression was evaluated via IHC (Fig. 4C). As expressed, decreased Ki-67 and increased TUNEL levels were observed following AZD1775 (90 mg/kg) monotherapy in HuCCT-1 and SNU2670 xenograft mice (Fig. 4C). Downregulation of Ki-67 and upregulation of TUNEL expression were maintained in combination treatment groups (Fig. 4C). In addition, p-ATR upregulation induced by AZD1775 was efficiently reduced by the combination regimen.
To confirm these observations, we detected cell proliferation and DNA damage markers such as p-AKT, p-ATR, WEE1, p-WEE1, and γ-H2AX. Consistent with the tumor growth curve and IHC results, AD1775 (90 mg/kg) alone dramatically blocked p-WEE1, WEE1, and p-AKT expression and upregulated γ-H2AX expression in both HuCCT-1 and SNU2670 xenograft mice (Fig. 4D). In the SNU2670 xenograft model, co-treatment with AZD1775 and AZD6738 dramatically upregulated γ-H2AX and downregulated p-AKT expression (Fig. 4D).
Discussion
Our study highlighted that ATM or ATR activation reduced the efficacy of WEE1 inhibition in BTC cells, and dual inhibition of ATR and WEE1 displayed potent anti-tumor effects both in vitro and in vivo.
In the current study, we focused on the resistance mechanism of AZD1775 and demonstrated that dual blockade of WEE1 and ATM or ATR has potent anti-tumor effects in BTC. AZD1775 reduced p-ATM and p-ATR levels in relatively sensitive cells, but in less sensitive cells, both p-ATM and p-ATR levels were elevated by AZD1775 (Fig. 3A). Another study described that WEE1 inhibition dramatically downregulated ATM expression in AZD1775-sensitive gastric cancer cells [16]. The other study reported that breast cancer cells were sensitive to WEE1 inhibition, and the IC50 was less than 1 μM in most cell lines [17]. However, they found that p-ATM and p-ATR expression was significantly upregulated by AZD1775 in a concentration- and time-dependent manner. Inconsistent with the other tumor types, WEE1 inhibition impairs p-ATM or p-ATR expression in BTC sensitive cells.
WEE1 inhibition could suppress cell migration in sensitive BTC cells (Fig. 2D). Although the potential anti-tumor effects of DDR-targeted agents have been evaluated in different cancer types, their effects on migration have not been well studied. Recently, in line with our data, other study reported that AZD1775 has an anti-migration/invasion effect in gastric cancer; however, the mechanism was not elucidated [18]. ATM is also associated with cell migration via interleukin-8 regulation [19]. As shown in Fig. 2D, AZD1775 did not impair the migration of less sensitive cells. It could be speculated that activated ATM or ATR facilitated cell migration even in the presence of WEE1 inhibition. Our result suggests that DDR core members have a critical role in cell migration.
Given the encouraging effects of DDR-targeted agents against multiple cancer types [1], the anti-tumor effect of these compounds urgently need to be evaluated in BTC.
In triple-negative breast cancer, WEE1 inhibition overcame cisplatin resistance by disrupting DNA replication and G2/M cell cycle checkpoint regulation [20]. A clinical trial also revealed that BTC patients harboring DDR-related gene mutations responded better to cisplatin therapy than those with wild-type cancer [21]. Because cytotoxic chemotherapy is the standard treatment for advanced BTC, targeting DDR signaling pathways is extremely important for improving the efficacy of chemotherapeutic agents against BTC. Recently, we demonstrated that ATMlow/p53low BTC cells were sensitive to ATR inhibitors, and combination treatment with cytotoxic chemotherapeutic agents synergistically induced DNA damage [10]. These data illustrated that BTC is a good candidate for implementing DDR-targeted agents.
Accumulating evidence has revealed that DDR signaling pathways modulate the tumor microenvironment via immune response networks in many cancer types [22,23]. WEE1 inhibition enhances the efficacy of programmed cell death-1–targeted treatment efficacy by amplifying cytotoxic T-lymphocyte killing ability [22]. Some studies also discovered that ATR inhibition-mediated downregulation of programmed cell death ligand-1 expression facilitated CD8+ T cell toxicity [23]. Based on these roles in immune modulation, we speculated that the combination of WEE1 and ATR inhibition might more potently activate T cell-dependent cytotoxicity in BTC. This should be further studied.
To date, co-targeting of WEE1 and ATR has rarely been investigated, although one study reported that this combination had a potent anti-tumor effect in triple-negative breast cancer [17]. The researchers demonstrated that Rad51 inactivation and γ-H2AX accumulation were more significantly observed after combination treatment. Notably, ATR inhibitor as single agents has been evaluated in HER2-positive breast cancer [24]. The investigators found that AZD6738 reduced several DNA repair proteins and p-AKT levels in sensitive cells but not in insensitive cells. The earlier studies have been demonstrated that AKT involves in DNA damage repair especially through non-homologous end joining pathway [25]. In our study, p-AKT level was blocked by AZD1775 or AZD6738 in both HuCCT-1 and HER2-positive SNU2670 xenograft models (Fig 4D). Although we have not further elucidated how AZD1775 or AZD6738 mediate p-AKT level, it is meaningful for additional investigation using this combination strategy in BTC.
Taken together, DDR-targeted treatment using WEE1 and ATR inhibitors exerted potent anti-tumor effects in both in vitro and xenograft models of BTC. These findings support further clinical development of the DDR-targeted strategy in patients with BTC. Our data indicated that DDR-targeted combination treatment could represent a potential treatment strategy for patients with BTC.
 XML Download
XML Download