

 PDF
PDF Citation
Citation Print
Print


Introduction
Myeloproliferative neoplasms (MPNs) are a group of disorders in which bone marrow (BM) stem cells show abnormal growth and reproduction. In MPNs, abnormal stem cells produce excess numbers of one or more types of blood cells. These abnormal cells do not function properly and can cause serious health problems unless properly treated and controlled. Chronic myeloid leukemia (CML) is a type of MPN characterized by the translocation between chromosome 9 and 22, which is called Philadelphia chromosome, thereby resulting in the BCR-ABL1 oncogene. According to ‘The 2016 revision to the World Health Organization classification of myeloid neoplasm and acute leukemia’, other classical MPNs can be diagnosed after CML is excluded. However, recent cases with the coexistence of BCR-ABL1 and other MPN markers (i.e., CALR, JAK2, or MPL mutations) have been reported. Herein, we present a case of BCR-ABL1–positive CML with a CALR mutation in a patient with persistent thrombocytosis who maintained a major molecular response after treatment with a tyrosine kinase inhibitor (TKI).
Case Report
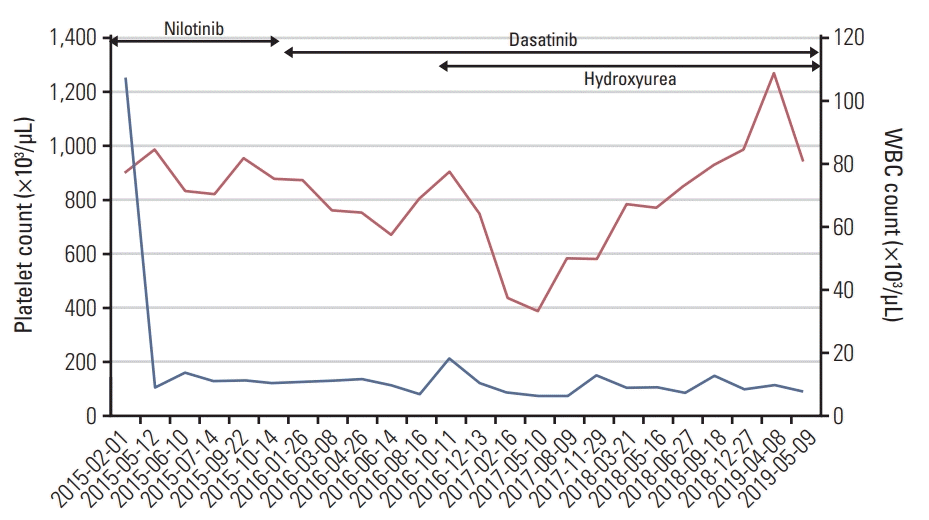
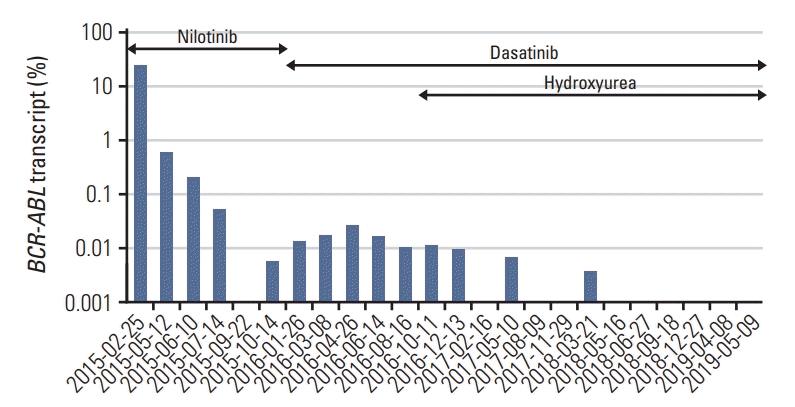
A 33-year-old woman was referred to our hospital in February 2015 owing to an abdominal mass and abnormal laboratory values. Abdominal ultrasonography revealed splenomegaly (22 cm), and laboratory examinations revealed a white blood cell (WBC) count of 107,600/mm3 and a platelet (PLT) count of 906,000/mm3. BM examination revealed a marrow cellularity of 90%, with markedly increased megakaryocytes. Reticulin staining indicated grade 2-3 fibrosis. The karyotype was 45,XX,t(9;22)(q34;q11.2),der(13;14)(q10;q10)[15]. In case of der(13;14)(q10;q10), it was found in all cells continuously after treatment and presumed to be germline mutation. Fluorescence in situ hybridization analysis showed that 99.2% of 500 cells were positive for t(9;22), and real-time quantitative polymerase chain reaction showed that the transcript level for BCR-ABL1/ABL1IS (international scale [IS]) was 25.603%. JAK2 V617F was negative on conventional screening. Testing for CALR mutations was not yet available. Accordingly, the patient started receiving nilotinib. After 6 months of treatment with nilotinib, she achieved a major molecular response. In October 2015, she developed persistent thrombocytosis. A repeat BM examination did not show any specific findings. The karyotype was 46,XX,der(13;14) (q10;q10)[13]. However, as thrombocytosis worsened (PLT count, 1,006,000/mm3) and splenomegaly persisted, we changed nilotinib to dasatinib. We then added hydroxyurea as part of the treatment, after which the thrombocytosis was controlled slightly but it worsened subsequently (Figs. 1 and 2). In May 2019, follow-up BM examination was performed, and we used the BM aspiration sample for MPN–next-generation sequencing (NGS): the patient had CALR mutation (c.1099_1136delinsAGGT), with an allele frequency of 15.97%, and TET2 mutation (c.3409+1G>A), with an allele frequency of 7.83%. We then retrospectively performed repeat MPN-NGS testing using the BM aspiration sample obtained in 2015. We found that CALR mutation had been presence since 2015, with an allele frequency of 10.64%, but no TET2 mutation was observed. We reported a case of CALR-positive CML with persistent thrombocytosis during TKI treatment.
The study protocol was approved by the Institutional Review Board of the Soonchunhyang University Seoul Hospital (No. 2020-01-012). The study patient was informed as the purpose of the study and provided her consent.
Discussion
CALR mutation was first recognized as a somatic mutation in patients with MPN who had no mutation in either JAK2 or MPL in 2013 [11]. CALR mutation is observed in approximately 70% of patients with essential thrombocythemia (ET) or primary myelofibrosis who did not carry a mutation in either JAK2or MPL[11,12]. CALR-mutant MPNs are characterized by a gene signature associated with activating JAK2 signaling, and CALR-mutants activate the JAK/STAT pathway through MPL, leading to excessive PLT production [13]. More than 50 different mutations in CALR have been described, but a 52 base-pair deletion (type 1) or a 5 base-pair insertion (type 2) account for more than 80% of mutations [13]. Pietra et al. [14] showed an association between CALR type 1 mutations and a significant increase in the risk of myelofibrotic transformation among ET cases, whereas type 2 muta-tions were preferentially associated with an ET phenotype, low risk of thrombosis despite a very high PLT count, and an indolent clinical course.
The presence of a BCR-ABL1 translocation with a coexisting mutation in CALRis exceptionally rare. A total of 10 cases have been reported since the first report was published in 2015, and the current report is the first in Korea (Table 1). In most cases, CALR mutation and BCR-ABL1 translocation were synchronously identified or BCR-ABL1 translocation was detected after CALR mutation. Otherwise, the presence of CALR mutation was not confirmed, but there was a case where the diagnosis was changed to CML on the basis of the findings of ET. To the best of our knowledge, there has been no report of CALR mutation after CML diagnosis. TKIs were ineffective for CALR-mutated clones, as shown in previous studies. Accordingly, physicians should be careful while changing the treatment of patients with CML who achieve a molecular response but do not obtain a hematologic response. Additional molecular mutations may be identified in patients with CML with the recent widespread use of NGS testing. A recent study showed the presence of molecular aberrations other than JAK2 V617F and BCR-ABL1 in 56% of patients with MPN, including mutations in TET2, ASXL1, RUNX1, CBL, DNMT3A, PHF6, SF3B1, and TP53[15]. In particular, TET2 is a putative tumor suppressor gene located on chromosome 4q24. Only a few studies revealed TET2 mutations in patients with MPN. However, there is no consensus regarding the exact role of TET2. In the current case, the relevance of identifying a new TET2 mutation during the course of the disease is unclear. Although patients with CML with BCR-ABL1 translocation have additional molecular aberrations, they are still diagnosed with CML on the basis of the current diagnostic criteria. However, further studies are needed regarding the diagnosis, treatment, and prognosis of these patients.
 XML Download
XML Download