

 PDF
PDF Citation
Citation Print
Print


Introduction
With the recent advances in molecular biology and its surrounding technology, the approach to diagnosis and treatment of cancers has dramatically changed [1]. Major cancer therapeutics involve a combination of cytotoxic drugs, but current treatment options are shifting toward molecular targeted agents [2,3] and immuno-oncology agents. To determine the appropriate treatment for individual patients, identification of appropriate biomarkers such as specific mutations or expression patterns are necessary. The liquid biopsy technology such as cell-free DNA (cfDNA) is becoming an increasingly popular source of non-invasive biomarkers for diagnosis and disease monitoring in cancer patients [4]. The cell-free circulating tumor DNA (ctDNA) was first reported in 1948 [5]. ctDNA has been evaluated for assessing metastasis, prognosis or diagnosis in breast, colorectal, lung cancer and various other neoplasms [6-11].
Epithelial ovarian cancer (EOC) is one of the more common cancers in women with more than 238,000 new cases diagnosed in 2012 worldwide [12]. Its mortality rate is highest among gynecologic cancers. With advancements of new systemic therapies and surgical techniques, the survival rate is improving. However, the total disease control remains poor because of eventual resistance to chemotherapy or other targeted drugs [13]. Mutation and loss of TP53 or BRCA1/2 function, or activation KRAS, BRAF, CTNNB1, and PIK3CA have been reported as genetic abnormalities in EOC [13]. KRAS and PIK3CA mutations are seen in around 10% of EOC. Since the hotspots of KRAS and PIK3CA mutations are well defined, analysis of those mutations represents a potentially sensitive and solid approach to evaluate ctDNA in the blood of EOC patients.
Reports of ctDNA for EOC have been increasing in the last few years. Most of the reports investigated TP53 mutation because it is the most frequently mutated gene in high-grade serous ovarian cancer (HGSC). TP53 mutation monitoring in cfDNA of HGCS patients showed potential as a biomarker for treatment response [14]. ctDNA can be detected even in some of the cancers in the early stage including EOC [15]. Exploratory analysis of PIK3CA or KRAS mutations in cfDNA of 29 ovarian clear cell carcinoma (OCCC) patients showed shorter progression-free survival (PFS) in the patients with detectable ctDNA [16]. These reports encourage the use of ctDNA as a tool for diagnosis, monitoring disease progression and response to treatment in EOC patients [14-21]. However, tumor-informed ctDNA for PIK3CA or KRAS mutations of other ovarian histotypes were unclear, and the analysis as to whether it is prognostic, particularly in early stage EOC, has yet to be established.
Our aim in this study is to evaluate mutation rates of KRAS or PIK3CA in the plasma of patients with an ovarian tumor harboring KRAS or PIK3CA mutations using droplet digital polymerase chain reaction (ddPCR) in the largest cohort thus far, and to analyze if there is an association between ctDNA status and clinicopathological features or clinical outcomes in EOC. In addition, we compared mutation frequency in the matched plasma collected at the time of initial diagnosis and recurrence in those patients.
Materials and Methods
1. Patients and sample collection
Patients with ovarian tumor who were treated in Saitama Medical University International Medical Center between 2010 and 2016 were included in this study. The clinicopathological data was retrieved from medical records. Patients with non-epithelial ovarian tumors, synchronous cancer and ovarian metastasis from non-gynecological origin were excluded from this analysis. A total of 306 ovarian tumor patients were included in the analysis.
Tumor specimens were collected from each patient at the time of initial surgery and stored at –80°C until use. Frozen tumor specimens from 306 patients with ovarian tumors were used for extraction of genomic DNA. In 306 patients, 226 (73.9%), 15 (4.9%), 14 (4.6%), 43 (14.1), and eight (2.6%) patients had EOC, fallopian tube cancer, primary peritoneal cancer, borderline tumor and benign tumor, respectively (S1 Table). We collected plasma from these patients before the surgery and stored at –80°C until use. In addition, we collected the plasma of 17 patients who suffered a relapse at the time of recurrence.
2. Tumor genomic DNA and plasma cfDNA extraction
Genomic tumor DNA was extracted from approximately 25-50 mg of frozen tumor sample using the NucleoSpin Tissue kit (Macherey-Nagel, Duren, Germany) according to the manufacturer’s instructions. Quantification of genomic DNA (gDNA) was performed using NanoDrop (Thermo Fisher Scientific, Waltham, MA). cfDNA was extracted from 0.5 mL of plasma (before surgery) and eluted in 60 μL of the supplied elution buffer using the QIAamp Circulating Nucleic Acid Kit (50) (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
3. Droplet digital PCR
ddPCR was performed using the PrimePCR for ddPCR PIK3CA E542K, E545K, H1047R or KRAS screening multiplex kit (G12A, G12C, G12D, G12R, G12S, G12V, G13D) (Bio-Rad catalog No. 186-3131, 186-3132, 186-3133 or 186-3506, Hercules, CA). KRAS screening multiplex kit screens seven KRAS mutations in a single well simultaneously. This kit cannot determine each KRAS mutation separately. Reactions were carried out in a reaction volume of 20 μL on a QX200 AutoDG Droplet Digital PCR System (Bio-Rad). The 20 μL PCR mix was composed of 10 μL 2×ddPCR supermix for probes (no dUTP), 1 μL of each (target and reference) 20×amplification primer/probe mix (450 and 250 nmol/L, respectively), 3 μL distilled water, and 5 μL gDNA or cfDNA extracted. The cycling conditions were as follows; initial denaturation at 95°C for 10 minutes, followed by 40 repeated cycles of 94°C for 30 seconds and 55°C for 60 seconds, a step of 98°C for 10 minutes and finally samples were maintained at 4°C. Results were analyzed with Quatasoft v1.7.4 (Bio-Rad). All gDNA samples were first evaluated by ddPCR and confirmed by other PCR based methods. As for cfDNA, we repeated each experiment using ddPCR at least twice. If there was a discordance, we performed the third run for confirmation. We defined the mutation as positive when we saw more than one copy of mutation by ddPCR (S2 Fig.).
4. Statistical analysis
The statistical analysis was conducted using JMP version 10 and GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA). Chi-square tests were used to estimate association between detection rate of mutation of ctDNA and clinicopathologial features. Survival analysis was performed by Kaplan-Meier methods and multivariate Cox regression models. Wilcoxon signed-rank test was performed to compare the level of cancer antigen 125 (CA125) and ctDNA at the time of recurrence and before the primary operation.
5. Ethical statement
This study was approved by the Institutional Review Board of Saitama Medical University International Medical Center (#14-058). Informed consents including future research purposes were obtained from all patients in the previous studies (#10-078 and #12-096), and the Institutional Review Board approved to use the research materials in the current study.
Results
1. PIK3CA and/or KRAS mutations in ovarian tumors
A total of 306 patients who had ovarian tumors was screened for somatic PIK3CA and or KRAS mutations using their tumor specimens. A consort diagram of all patients in this study is shown in Fig. 1. Two hundred and fifty-five, 43 and eight patients had EOC, borderline ovarian tumor and benign ovarian tumor, respectively. Among EOC patients, 89 (34.9%), 82 (32.2%), 60 (23.5%), seven (2.7%), and 17 (6.7%) patients had clear cell carcinoma, high-grade serous carcinoma, endometrioid carcinoma, mucinous carcinoma and other histotypes, respectively.
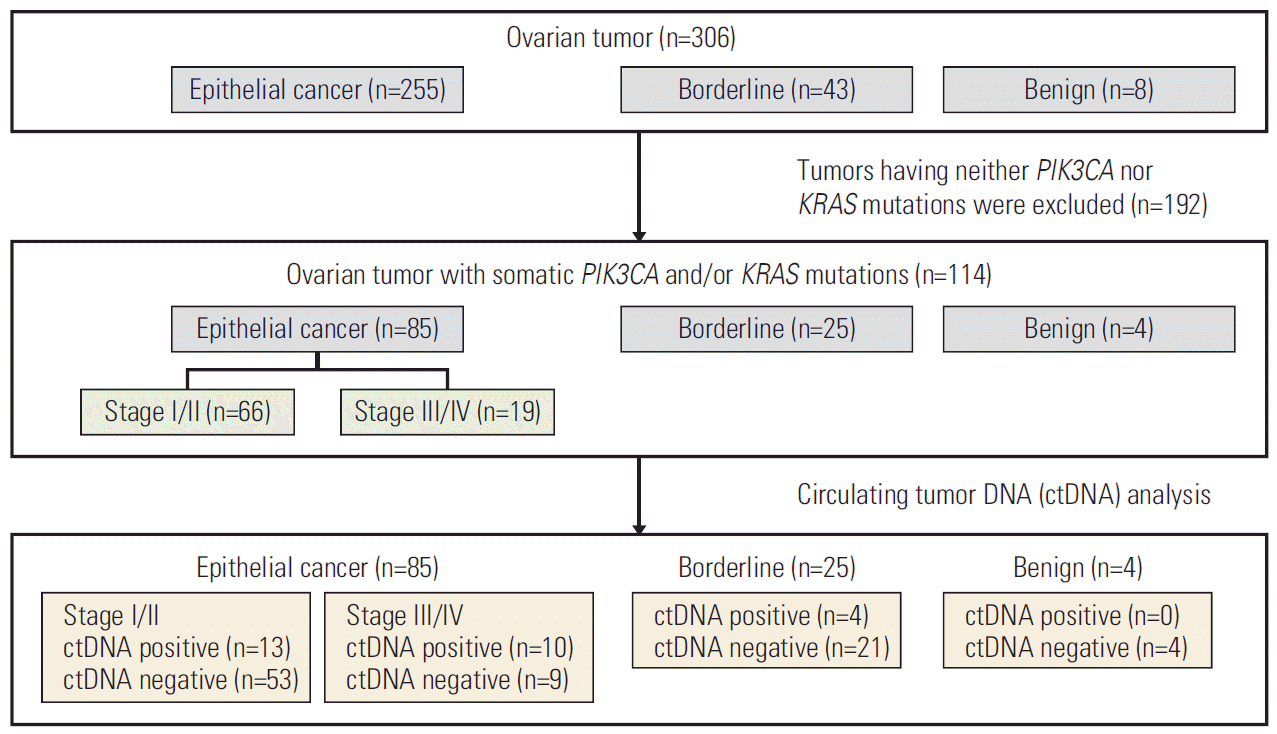
We found that 114 patients out of 306 patients (37.3%) had tumors with somatic PIK3CA and/or KRAS mutations using ddPCR as described in the materials and methods section (Fig. 1). Those mutations were found in 85/255 (33.3%), 25/43 (58.1%), 4/8 (50.0%) of EOC, borderline, and benign tumor, respectively (Fig. 1). In 255 EOC patients, somatic PIK3CA mutations were observed in 40/255 patients (15.7%), and KRAS mutations were observed in 48/255 patients (18.8%) (Table 1). Three cases (1.2%) had both PIK3CA and KRAS mutations.
Table 1.
Detection rates for circulating tumor DNA (ctDNA)
![]()
2. PIK3CA and/or KRAS mutations in plasma circulating tumor DNA
Next, cfDNA from the plasma of those 114 patients who had tumor PIK3CA and/or KRAS was tested for the corresponding mutations using ddPCR. We defined ctDNA detection to be positive when the corresponding mutations were detected in the plasma cfDNA. As shown in Table 1 and S3 Table, positive ctDNA was found in 27.1% (23/85), 16.0% (4/25), and 0% (0/4) of patients with EOC, borderline and benign tumor, respectively. Each PIK3CA or KRAS mutation was detected in 11/40 (27.5%) and 12/48 (25.0%) of cfDNA in EOC patients, respectively. Each PIK3CA or KRAS mutation was detected in 0/2 (0.0%) and 4/24 (16.7%) of cfDNA in borderline ovarian tumor patients, respectively. KRAS mutation was detected in 0/4 (0.0%) of cfDNA in benign ovarian tumor patients (Table 1).
3. Relationship between circulating tumor DNA and clinicopathological factors in EOC patients
We included only EOC patients in the subsequent analyses. We investigated the relationship between ctDNA status (positive or negative) and clinicopathological features in 85 EOC patients with PIK3CA and/or KRAS mutations (Table 2). High detection rate of ctDNA was associated with advanced stage and positive peritoneal cytology (p=0.008 and p=0.007, respectively), but not with other factors such as age, histologic type and status of residual tumor at primary surgery (Table 2).
Table 2.
Relationship between ctDNA (PIK3CA and/or KRAS) detection and clinicopathological features in EOC patients
![]()
4. Circulating tumor DNA and outcomes
Next, we examined a potential association between ctDNA status and patient outcomes. As shown in Fig. 2A and B, we observed that the ctDNA detection was associated with both shorter PFS and overall survival (OS) in EOC patients (p < 0.001 and p=0.017, respectively). Similar trends were observed when we separately analyzed the mutations for PIK3CA or KRAS. The PFS was significantly shorter in patients with PIK3CA or KRAS mutations in cfDNA (p=0.008 and p=0.004, respectively) (Fig. 2C and E). However, ctDNA mutations for PIK3CA or KRAS showed no significant difference for the OS (p=0.118 and p=0.072, respectively) (Fig. 2D and F).
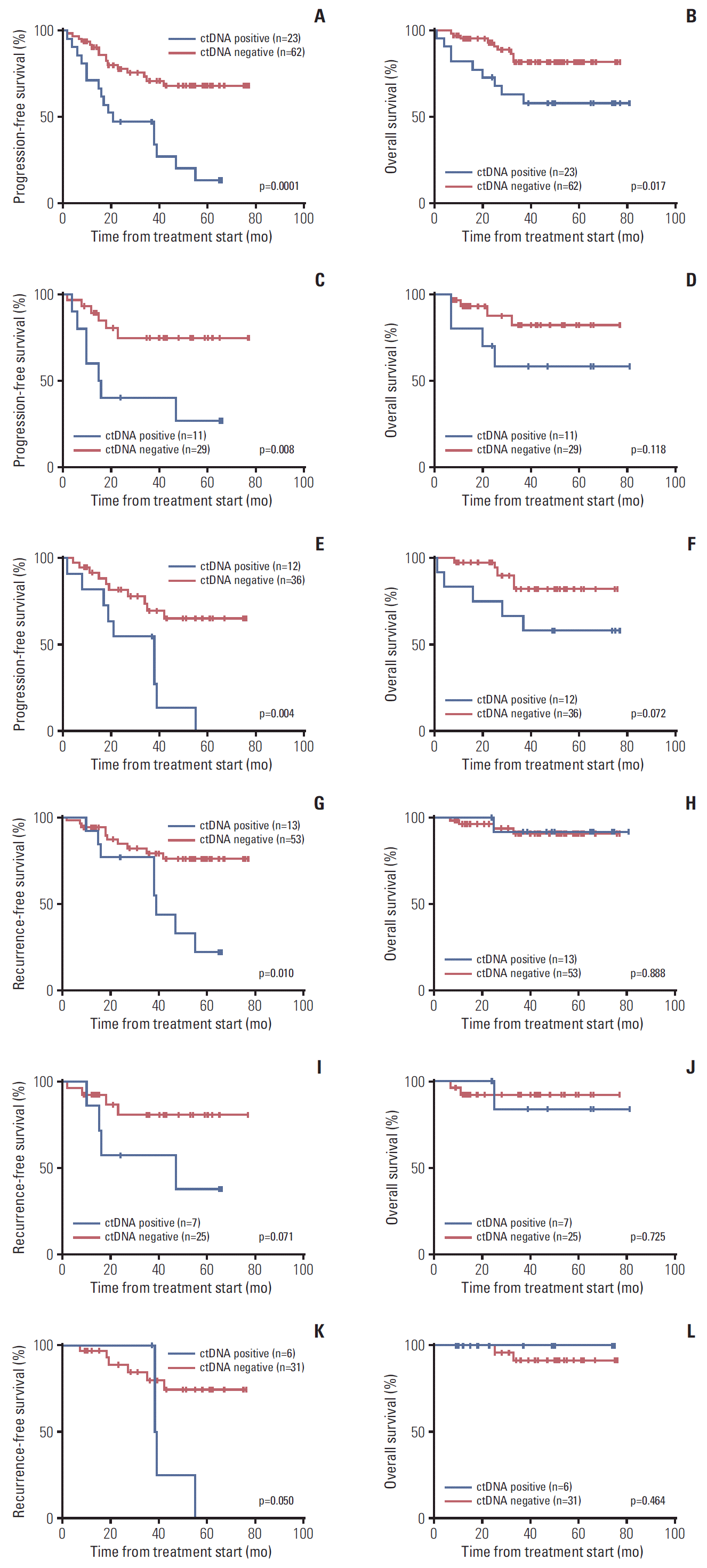
Fig. 2.
Survival curves according to circulating tumor DNA (ctDNA) status, positive or negative. Progression-free survival (A) and overall survival (B) in all epithelial ovarian cancer (EOC) patients as to ctDNA status for PIK3CA and/or KRAS mutations (p=0.0001 and p=0.017, respectively). Progression-free survival (C) and overall survival (D) in all EOC patients as to ctDNA status for PIK3CA mutations (p=0.008 and p=0.118, respectively). Progression-free survival (E) and overall survival (F) in all EOC patients as to ctDNA status for KRAS mutations (p=0.004 and p=0.072, respectively). Recurrence-free survival (G) and overall survival (H) in stage I/II EOC patients as to ctDNA status for PIK3CA and/or KRAS mutations (p=0.010 and p=0.888, respectively). Recurrence-free survival (I) and overall survival (J) in stage I/II EOC patients as to ctDNA status for PIK3CA mutations (p=0.071 and p=0.725, respectively). Recurrence-free survival (K) and overall survival (L) in stage I/II EOC patients as to ctDNA status for KRAS mutations (p=0.050 and p=0.464, respectively).
![]()
Additionally, we performed cox univariate analyses to assess the prognostic factors in those patients. We found that the ctDNA detection, stage and residual tumor status at the time of primary surgery were significant prognostic factors for PFS (p < 0.001, p < 0.001, and p < 0.001, respectively) (Table 3). Those factors were also significant prognostic factors for OS (p=0.012, p < 0.001, and p < 0.001, respectively) (Table 3). We further examined the multivariate analyses including ctDNA status, stage, and residual tumor status at the time of primary surgery, and histology. As shown in Table 3, ctDNA status, stage, residual tumor status at the time of primary surgery, and age remained as independent prognostic factors for PFS (p=0.010, p=0.001, p=0.006, and p=0.010, respectively). However, only histology, stage and age remained independent prognostic factors for OS (p=0.006, p < 0.0001, and p=0.041, respectively).
Table 3.
Cox regression models for overall and progression-free survival in EOC patients
![]()
5. A subgroup analysis of ctDNA in early stage EOC
As shown in Table 2, more than 50% of the tumor PIK3CA and/or KRAS mutations were found in early stage EOC, and we think that ctDNA may reflect potential tumor spread even when the tumor is clinically localized. Therefore, we analyzed ctDNA status in a subgroup of early stage (stage I-II) EOC patients. We found that 13 of 66 EOC patients (19.7%) who had stage I-II tumor were positive for ctDNA (Table 1). We found no association between ctDNA detection and any clinicopathological features in early stage EOC patients (age, International Federation of Gynecology and Obstetrics stage, histological type, and peritoneal cytology) (S4 Table). As shown in Fig. 2G, detection of ctDNA was associated with shorter recurrence-free survival (RFS) in early stage EOC patients (p=0.010, log-rank test). PIK3CA-mutant and KRAS-mutant ctDNA in early stage EOC patients showed trends toward a shorter RFS (p=0.071 and p=0.050, respectively) (Fig. 2I and K). However, no statistical difference was found in OS (p=0.725 and p=0.464, respectively) (Fig. 2J and L). We performed Cox univariate analyses to assess the prognostic factors including ctDNA status, stage, peritoneal cytology at the time of primary surgery, age and histology in early stage EOC patients. We found that ctDNA detection, peritoneal cytology at the time of primary surgery and age were significant prognostic factors for RFS in univariate cox regression models (S5 Table). However, in early stage EOC patients, we found no specific prognostic factors for OS in univariate cox regression models (S5 Table) and for both RFS and OS in the multivariate cox regression analysis (S6 Table).
6. Comparing ctDNA in paired plasma samples at primary and recurrent diagnosis in EOC patients
Tumors may change their molecular status during a relapse, metastasis or following chemotherapy-induced selection pressure. We compared ctDNA status in 17 paired plasma samples collected at the time of initial diagnosis and first recurrence. Detailed patient information and ctDNA status are described in S7 Table. Eight patients (47.1%) were ctDNA positive at the time of initial diagnosis, and seven of those eight patients (87.5%) remained ctDNA positive at the time of their first recurrence. We observed that one patient was ctDNA negative at the time of initial diagnosis but it became positive during recurrence. As shown in Fig. 3A, we did not observe any difference in the amount of cfDNA in the primary and recurrent tumor. The median total cfDNA (included mutation and wild-type) was 1,944 (range, 1,080 to 29,484 copies/mL) and 1,992 (range, 386 to 118,092 copies/mL) in patients with primary and recurrent tumors, respectively. When we examined the mutation rate in cfDNA, six of 17 patients (35.3%) had higher mutation rates in cfDNA at the time of recurrence compared to that at the time of primary diagnosis. However, no statically change was observed (Fig. 3B). In contrast, the levels of serum CA125 was lower and the tumor size was smaller when the patients had a recurrent tumor compared to that at the time of primary diagnosis (p < 0.0001) (Fig. 3C and D).
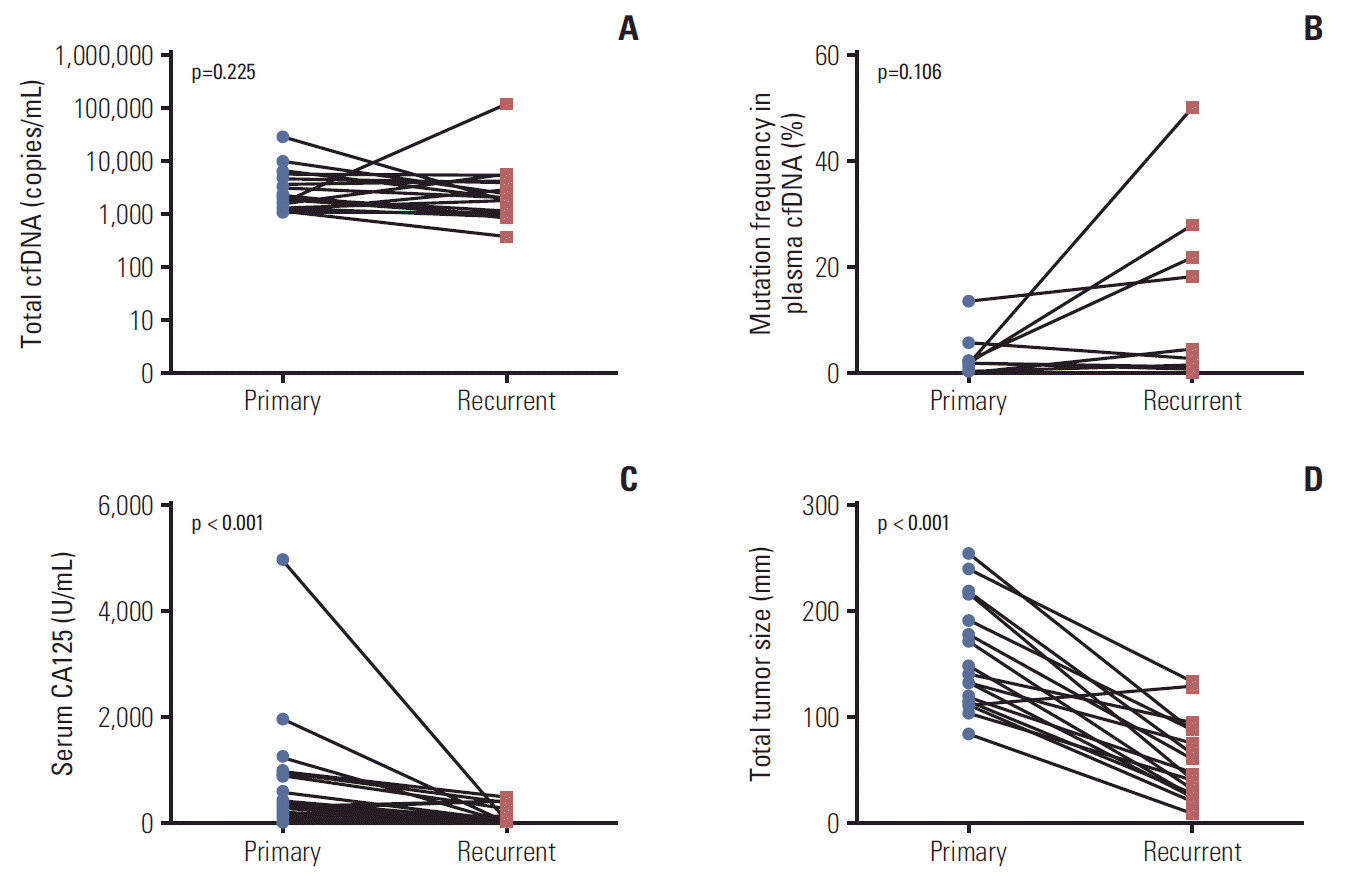
Fig. 3.
Quantitative analysis of circulating tumor DNA (cfDNA) at the time of primary treatment and at recurrence. (A) Total mutation copies in plasma. (B) Mutation frequency in cell-free DNA (cfDNA) ([mutated copies/wild type copies]×100). (C) Levels of serum cancer antigen 125 (CA125). (D) Tumor size.
![]()
Discussion
In this study, we evaluated ctDNA status in EOC patients by investigating PIK3CA and KRAS mutations in the plasma cfDNA using ddPCR, and found ctDNA detection in 27.1% of EOC patients, 19.7% in early stage and 52.6% in advanced stage patients. The presence of ctDNA in the blood at the time of primary treatment was an independent prognostic factor for recurrence or relapse in EOC patients. In addition, increased mutation rates in cfDNA was observed in patients with recurrence compared with those at primary treatment.
There are some reports of tumor-informed mutations in cfDNA of EOC patients [14,16-20,22]. However, most of the studies have addressed a relatively small EOC patient cohort. Because of a high frequency of mutations in the TP53 gene in HGSC patients, most of the reports for EOC previously examined TP53 mutations in plasma cfDNA [14,17-20]. The first report of tumor-specific mutations in cfDNA for EOC evaluated TP53 gene mutations, which were examined by fluorescence-based single-strand conformation polymorphism. Forty-four percent (12/27) of the patients with EOC were found to have a TP53 mutation in the tumor, and 16.7% (2/12) of those patients had matched mutations in the plasma cfDNA [19]. Morikawa et al. [16] investigated PIK3CA and KRAS mutations in 29 cases of OCCC. Eight of 29 patients (27.6%) had PIK3CA and/or KRAS mutations, and they observed ctDNA in three of those (37.5%) patients by ddPCR [16]. KRAS and PIK3CA mutations in cfDNA were also evaluated in endometrial cancer by targeted sequencing, and 14% (2/14) and 33% (7/21) of the tumors had KRAS and PIK3CA mutations in cfDNA, respectively [23]. Our study demonstrated ctDNA detection in 27.1% of plasma cfDNA of EOC patients using ddPCR for KRAS and PIK3CA mutations. It is mostly in agreement with previous studies, and is the largest sample size assessing detection rate of plasma cfDNA in EOC patients.
We observed that ctDNA detection was associated with advanced stage and positive peritoneal cytology (p=0.008 and p=0.007, respectively). In 40 patients with HGSC, the levels of ctDNA (AC/mL) for TP53 mutations correlated with tumor volume measured using 3D volume reconstructing from computed tomography (CT) images [20]. However, the other report for EOC showed no correlation between ctDNA status and other clinical and pathological factors in 69 EOC patients [17]. This discrepancy might be attributed to the fact that their study included only HGSC patients mostly in advanced stage disease, unlike our study that included all histotypes. When other types of cancers we examined, the clinical stage was reported to be the only clinical factor that affected the detection rate of ctDNA in colorectal cancer patients [24]. Higher performance status, presence of bone metastasis and metastasis in three or more organs were associated with high ctDNA detection rate in lung adenocarcinoma patients who had epidermal growth factor receptor (EGFR) driver mutations [25].
We demonstrated that presence of ctDNA in the blood at the time of initial treatment was found to be a prognostic factor for PFS and OS in EOC patients harboring PIK3CA and/or KRAS somatic mutations. Our data are mostly in agreement with previous preliminary reports by other investigators. TP53, PIK3CA, or KRAS mutations in cfDNA were associated with poorer survival in ovarian cancer [16-18,20,21], but they included a limited sample size in those analyses. Previous reports have shown that not only in EOC, but also other cancer types including endometrial, breast, colon and pancreatic cancer positive for ctDNA, was also associated with worse prognosis [3,18,26,27]. Our study demonstrated for the first time that ctDNA detection in plasma at the time of initial treatment was an independent prognostic factor by multivariate cox analysis in EOC patients. However, ctDNA was not shown to be an independent prognostic factor for OS in this study. This might be in part due to the complex treatments for recurrent EOC patients or limited events for multivariate analysis on OS in this study. We also investigated the role of ctDNA in each histotype. We found both PFS and OS were decreased in patient with clear cell carcinoma who had ctDNA compared to those who did not (S8A and S8B Fig.). The PFS but not the OS was decreased in the patients with endometrioid carcinoma who had ctDNA compared to those who did not (S8C and S8D Fig.). In contrast, no difference was found in either PFS or OS for serous and mucinous carcinoma. However, the numbers of cases in those two histologic types were limited (S7E-S7H Fig.).
This is the first report comparing paired plasma samples at the time of primary surgery and recurrence for ctDNA evaluation. The levels of serum CA125 and the tumor size decreased at the time of relapse than those at the time of primary treatment. Interestingly, we found a higher mutation rate in plasma cfDNA at the time of recurrence compared with at the time of primary treatment though statistically not significant. This may be partly explained by the different molecular status between the primary and relapsed disease. PIK3CA or KRAS mutant clones may be enriched during the relapse, suggesting a more aggressive phenotype with PIK3CA/KRAS mutants. By monitoring plasma ctDNA, we may detect ovarian cancer relapse earlier. Other reports also showed that ctDNA was detected in six of 44 patients with ovarian or endometrial cancer, even when CT scanning was negative for those tumors [18]. A few cases of OCCC were shown to have increased ctDNA earlier than CA125 at the time of relapse [16].
Our study has several limitations. One is the retrospective design and the second is modest sample size and all cases arising from single institute, and specifically studying cases with PIK3CA and KRAS mutations without studying TP53 mutations, which might affect the survivals in HGSC patients. The limited amount of plasma used in this study might affect the detection rate. Further studies using a large sample size or prospective design are warranted.
In conclusion, plasma ctDNA was detected in approximate 30% of EOC patients at the time of initial treatment in this study. The presence of ctDNA in the blood was shown to be an indicator for outcomes in EOC patients, suggesting that the presence of ctDNA could predict tumor spread even in cases of localized tumors.
 XML Download
XML Download