

 PDF
PDF Citation
Citation Print
Print


Introduction
Lung cancer is still the most common malignant tumor (11.6% of total new cancer cases) and the leading cause of cancer mortality (18.4% of the total cancer deaths) worldwide in 2018 [1]. Non-small cell lung cancer (NSCLC) is the most common pathological type, accounting for about 84% [2]. Nowadays, many proteins have been found to play a specific role in carcinogenesis and development of NSCLC, and some of them can be used as therapeutic targets. Finding a proper candidate therapeutic target for NSCLC to improve the prognosis of patients is still the research focus.
Caspase recruitment domain containing protein 9 (CARD9), a member of caspase recruitment domain family, has been found to be involved in many physiological processes including innate immune response, inflammation, and carcinogenesis [3-5]. Recently, abnormal expression of CARD9 has been reported in various types of malignant diseases. The elevation of CARD9 mRNA in gastric B-cell lymphoma [6] indicated that CARD9 was related with tumorigenesis and development. CARD9 can promote tumorigenesis of large intestines in the male APCmin mice, which mimics the genetic lesions associated with human familial adenomatous polyposis [7]. In addition, upregulation of CARD9 was also found in renal cell carcinoma and was significantly associated with poor prognosis [8]. These studies demonstrated a devil role of CARD9 in tumor development.
However, CARD9 appears to function differently in lung cancer. Our previous research on the difference of protein profiles of the benign and malignant pleural effusion by proteomics technology found that CARD9 was decreased in malignant pleural effusion, and the mechanism was not clear [9]. In the latest study of CARD9 in lung cancer, Qu et al. [10] reported that CARD9 suppressed lung cancer development by inhibiting the expansion of myeloid-derived suppressor cells (MDSCs) and indoleamine 2,3-dioxygenase (IDO) production.
In our study, we found that CARD9 expression was reduced in NSCLC tissues and low CARD9 expression was significantly associated with poor prognosis of NSCLC patients. We also identified the location of CARD9 in freezing section of clinical NSCLC specimen and found that CARD9 was expressed both in tumor cells and macrophages. Meanwhile, considering the different CARD9 baseline in various NSCLC cell lines, we speculated tumor-cells-derived CARD9 may bear some function in tumorigenesis. In addition, we investigated the role of CARD9 in NSCLC cells. By generating CARD9-overexpressing and CARD9-knockdown NSCLC cell lines in vitro, we observed that CARD9 could promote cell apoptosis and inhibit cell cycle progression, invasion, migration via mitogen-activated protein kinases (MAPK)/p38 signaling pathway. And coimmunoprecipitation (Co-IP) result showed that B-cell lymphoma 10 (BCL10) interacted with CARD9. It was implied that CARD9 may work differently in NSCLC tumorigenesis, progression and metastasis, which may be a new potential target for NSCLC therapy.
Materials and Methods
1. Patient specimens
Human specimens containing 94 cases of NSCLC tissues with adjacent normal lung tissues (> 5 cm from the lesion) for immunohistochemical analysis were collected from February 2013 to December 2014. Fresh surgical specimens of NSCLC tissues and paired adjacent normal lung tissues for testing CARD9 expression and location by western blot, polymerase chain reaction (PCR), and immunofluorescence were collected from July 2018 to December 2018. All these samples were obtained from the Huadong Hospital, Fudan University. The inclusion criteria were as follows: patients with primary lung cancer without other tumor diseases; all patients who underwent radical resection had no chemotherapy or radiotherapy before surgery; absence of cancer within 5 years before surgery. Patients without accurate medical records were excluded from this study. The specimens were staged according to the American Joint Committee on Cancer and the histological type were classified according to the World Health Organization classification. Paraffin blocks containing formalin-fixed specimens were available for histological examination and immunohistochemistry (IHC) from 94 NSCLC tissues and paired adjacent normal lung tissues. The follow-up began from the date of surgery and the deadline was April 1, 2017. Overall survival (OS) was defined as the time from surgery to death or from surgery to the deadline for the surviving patients. Progression-free survival (PFS) was defined as the time from surgery to the first progression or death.
2. Immunohistochemistry
All sections were heated at 60℃ for 30 minutes and then de-waxed in xylene and rehydrated in a series of graded ethanol. The endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 minutes and subsequently microwave antigen retrieval was performed with ethylenediaminetetraacetic acid buffer (1 mmol/L, pH 8) for 30 minutes. After being blocked with the goat normal serum for 10 minutes, the sections were incubated with rabbit monoclonal anti-CARD9 antibody overnight at 4℃ followed by incubation with horseradish peroxidase (HRP)–conjugated goat anti-rabbit secondary antibody for 1 hour at room temperature. The immunostaining was performed by 3,3-diaminobenzidinetetrahydrochloride and Mayer’s hematoxylin counterstain was applied. Negative controls consisted in the absence of primary antibody.
3. Evaluation of immunostaining
The level of CARD9 expression was performed by semiquantitative analysis as described previously [11]. The intensity of immunostaining was evaluated by the degree of color and then was scored as none (0), weak (1), moderate (2), and strong (3). The distribution of positive tumor cells was defined as follows: 0, 0%; 1, 1%-20%; 2, 21%-40%; 3, 41%-60%; 4, 61%-80%; and 5, 81%-100%. Both of them were evaluated by two independent observers who were unaware of the clinicopathologic features of the patients. The expression of CARD9 in all samples were finally scored by multiplying the intensity and the percentage ranging from 1 to 9. The immunohistochemistry (IHC) scores of CARD9 expression in NSCLC tissues were divided into two levels: low (0-3) and high (4-12).
4. Immunofluorescence
The fresh tissues were frozen in tissue freezing medium at –80℃ immediately and was sliced into 4 μm section for immunofluorescence (Leica CM1950, Jena, Germany). Briefly, frozen tumor sections were fixed by Immunol Staining Fix Solution (Beyotime, Shanghai, China) at room temperature for 10 minutes and washed by phosphate buffered saline (PBS). Then it was blocked with PBS solution containing 5% goat serum and 0.2% Triton X-100 at room temperature for 30 minutes and incubated with primary antibody mixture overnight at 4℃. Anti-CD68 was used to stain macrophages, and anti–epithelial cell adhesion molecule (EpCAM) was used to stain NSCLC cells (S1 Table). After washing by PBS, freezing slices were incubated with fluorescent secondary antibody mixture at room temperature in dark for 1 hour followed by three times PBS washing. DAPI was used for nuclei staining (Beyotime) for 5 minutes. After washing, slices were covered by Antifade Mounting Medium (Beyotime) and coverslip. Confocal laser scanning microscope (Leica TCS SP8) was used for detection.
5. Cell culture and transfection
Human cell lines (human bronchial epithelioid cells [HBE], H1299, A549, 95C, 95D, PC9, and THP-1) were obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China), and BEAS-2B cells (human normal lung epithelial cells) was obtained from Beyotime. All these cells were maintained in Roswell Park Memorial Institute-1640 (RPMI-1640, Hyclon, Logan, UT) supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA) at 37℃ in a humidified atmosphere containing 5% CO2. To establish stable CARD9 silencing cell lines, cells were transfected with GFP/CARD9 ShRNA lentivirus construct (GeneChem, Shanghai, China) (shCAR-D9 group) while cells transfected with the empty lentivirus vectors (shControl group) were considered as a negative control. The stable cell lines were selected with 5 μg/mL puro-mycin (Beyotime) for 1 week and the downregulated level of CARD9 were verified by quantitative realtime PCR (qRT-PCR) and western blot. CARD9 gene was ligated into the FV115 plasmid (Huzbio Technology, Shanghai, China), which was used to generate CARD9 overexpression cell lines. Firstly, 1×106 cells were seeded into 6-well plate, which were transfected with FV115-CARD9 or FV115 plasmid with instruction of Lipofectamine 6000 (Beyotime) after 24 hours. Stably transfected cell lines were selected by puromycin and verified by real-time quantitative reverse transcription polymerase chain reaction and western blot. In the inhibition assay, SB203580 (Cell Signaling Technology, Danvers, MA) dissolved in dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO) was added to culture medium when cells density rea-ched to 50%-70%. The final concentration of SB203580 was 10 nM. Same volume of DMSO was added as the control. Two hours later, cells were re-cultured in the fresh RPMI-1640 or collected for further experiments.
6. Western blot analysis
The total cellular proteins were extracted with RIPA buffer (Beyotime). Equal amounts of protein (30 μg/lane) were separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA). After blocking with PBS buffer (Beyotime) containing 5% non-fat milk and 0.1% Tween 20 (Beyotime), membranes were incubated with primary antibody over-night at 4℃. Subsequently HRP-conjugated secondary antibodies were incubated for 1 hour at room temperature and developed with enhanced chemiluminesence (Beyotime). All antibodies used were listed in S1 Table.
7. Quantitative real-time PCR analysis
Total cellular RNA was isolated with TRIzol reagent (Beyotime) following the manufacturers’ instructions. Equal amounts of RNA (1 μg) were used as template to synthesize first-strand cDNA with the PrimeScript RT reagent Kit with gDNA Eraser (Takara, Dalian, China). Then each PCR reac-tion contained a total volume of 20 μL SYBR Premix Ex Taq (2 μL cDNA, 0.4 μL of each specific primer, 0.4 μL of ROX Reference Dye [50×], 10 μL SYBR Premix Ex Taq, and 6.8 μL RNase free water, Takara) was performed with an Applied Biosystems 7500 Fast Real-Time PCR System (Foster City, CA, USA) at the following conditions: 95℃ for 30 seconds, 40 cycles at 95℃ for 15 seconds, and 60℃ for 30 seconds. β-actin was used as an internal control to normalize the expression of target gene. The PCR reaction for each sample was done in triplicate and the experiments were performed for 3 times. The relative quantity of target gene expression was calculated according to 2-ΔΔCT method. Primers used in this study were as follows: CARD9: F, ATGTCGGACTACGAGAACGAT; R, TGATGCGTGAGGGGTCGAT; β-actin: F, GTGGACATCCGCAAAGAC; R, AAAGGGTGTAACGCAACTA.
8. Cell growth assay and clonal formation assay
Cell growth assays were performed following the manufacturer’s instruction to test the cell viability. Cells were seeded at a density of 2×103/well into the 96-well plates and then checked at 0, 24, 48, 72, and 96 hours by the Cell Counting Kit-8 (Beyotime) and the Synergy H1 microplate reader (BioTek, Winooski, VT) at 450 nm. As for clonal formation assay, following transfection and selection, 200 cells were seeded in 6-well plate in triplicates and incubated for 7 days. Then cells were fixed with 10% ice-cold methanol and stained with 0.5% crystal violet solution. Colonies consisting more than 50 cells per colony were counted. Independent experiments were conducted triplicates.
9. Cell cycle and apoptosis analysis
For cell cycle analysis, cells were starved for 12 hours before trypsinization (Beyotime). Trypsinizated cells were washed by cold PBS and centrifuged at 2,000 rpm for 5 minutes. Fifty microliters of PBS was used to resuspend the cells gently, and then 950 μL 75% ethanol was added to fix the cells at 4°C overnight. The fixed cells were washed with cold PBS, centrifuged at 2,000 rpm for 5 minutes and incubated with 500 μL PI/RNase Staining Buffer (BD Biosciences, Franklin Lakes, NJ) at room temperature in dark for 15 minutes. Before analysis, cells were sieved by 200 mesh sieves. For cell apoptosis analysis, cold PBS-washed cells were resuspended by 1× binding buffer and then every 1×105 cells were incubated with 5 μL Annexin-V/7-ADD (BD Biosciences) double staining at room temperature in dark for 30 minutes. Established cells above were tested by flow cytometry (BD FACSAria II). Flow jo 10 and Modfit LT were used to analyze cell apoptosis and cell cycle, respectively.
10. Wound healing assay and Matrigel invasion assay
For wound healing assay, cells were seeded in 6-well plates with 5×106/well. When they grew to 90% confluence, the cell monolayer was scratched with a sterile 200 μL pipette tip. The detached cells were removed by washing with PBS and then the cells were maintained in medium containing 1% FBS and 2 nM Thymidine (CSNpharm, Arlington Heights, IL). Cells migrated to the wounded area were photographed at 0 hour and 24 hours using microscopy (Nikon ECLIPSE Ti, Tokyo, Japan). The experiments were performed in triplicate. For the invasion assay, 50 μL Matrigel (BD Biosciences) was precoated on the upper chambers (Corning Costar, New York, NY) and then 2×104 cells with 200 μL serum-free medium were added. Five hundred microliters of medium containing 10% FBS was added to the lower chambers. After 24 hours of incubation at 37℃, cells on the top of the membrane were removed and cells on the bottom of the membrane were fixed with 10% ice-cold methanol and stained with 0.5% crystal violet. Chambers placed on the glass slide were photographed under microscopy (Nikon ECLIPSE Ti). All of the cells under the chambers were counted without overlapped horizons. The experiments were performed in triplicate.
11. Coimmunoprecipitation
Cell lysates from successfully transfected H1299 cells or PC9 cells were collected and immunoprecipitated with either 5 μg anti-CARD9 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or 1 μg anti-mouse IgG antibody (Beyotime) at 4℃ overnight. The cell lysates were then added with 30 μL protein A/G magnetic beads (MedchemExpress, Monmouth Junction, NJ) and rotated for at 4℃ overnight. After washing by PBS containing 0.5% Triton X-100 for 5 times, beads were collected in 15 μL 1× SDS loading buffer and boiled for 5minutes. Then protein samples in SDS were collected for immunoblotting as we described above. HRP-mouse anti-rabbit IgG heavy chain specific was used as the secondary antibody to BCL10 antibody to exclude the influence of light chain.
12. Statistical analyses
Statistical analyses and graphs were performed by SPSS ver. 25.0 software (IBM Corp., Armonk, NY) and Prism ver. 7.00 (GraphPad Software, San Diego, CA). The results of Western blot were quantified by Image J (National Institutes of Health, Bethesda, MD). Data under the banks presented as the mean of three results of Western blot. IHC score of CARD9 expression in NSCLC tissues and adjacent normal tissues were analyzed by Wilcoxon rank-sum text. The relationship between CARD9 expression and clinicopathological features was analyzed by the chi-square test and Fisher exact test. The survival curves were made by the Kaplan-Meier method and analyzed by the log-rank test. Binary logistic regression models were used to evaluate the independent prognostic factors. The differences between vector groups and CARD9 groups, shControl groups, and shCARD9 groups were analyzed by Student’s t test. p < 0.05 was considered statistically significant in all the cases.
Results
1. CARD9 was downregulated in NSCLC tissues
To investigate the expression of CARD9 in NSCLC, we first analyzed the difference of CARD9 mRNA between NSCLC tissues and adjacent normal lung tissues using The Cancer Genome Atlas (TCGA) database. Both in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), CARD9 mRNA was decreased in tumor tissues compared with paired normal lung tissues (Fig. 1A, p=0.001; Fig. 1B, p < 0.001). To confirm the results, we examined our cohort consisting 94 patients with NSCLC by IHC. CARD9 was mainly located in cell the nucleus and cytoplasm, and IHC score showed CARD9 in carcinoma tissues was lower than in adjacent normal tissues (p < 0.001) (Fig. 1C and D) both in LUAD and LUSC. Consistently, by testing surgical lung resection specimens, CARD9 was found to be decreased in lung tumor tissues in comparison with normal tissues at protein (Fig. 1E) and mRNA levels (p=0.039) (Fig. 1F). CARD9 has previously been identified to be expressed in myeloidderived cells such as macrophages, dendritic cells and MDSCs. To confirm the role of CARD9 in lung tumorigenesis, we used immunofluorescence to testify the cellular location of CARD9. As shown in Fig. 1G, despite weak expression, we can still observe positive CARD9 expression in macrophages (anti-CD68) and lung tumor cells (anti-EpCAM).
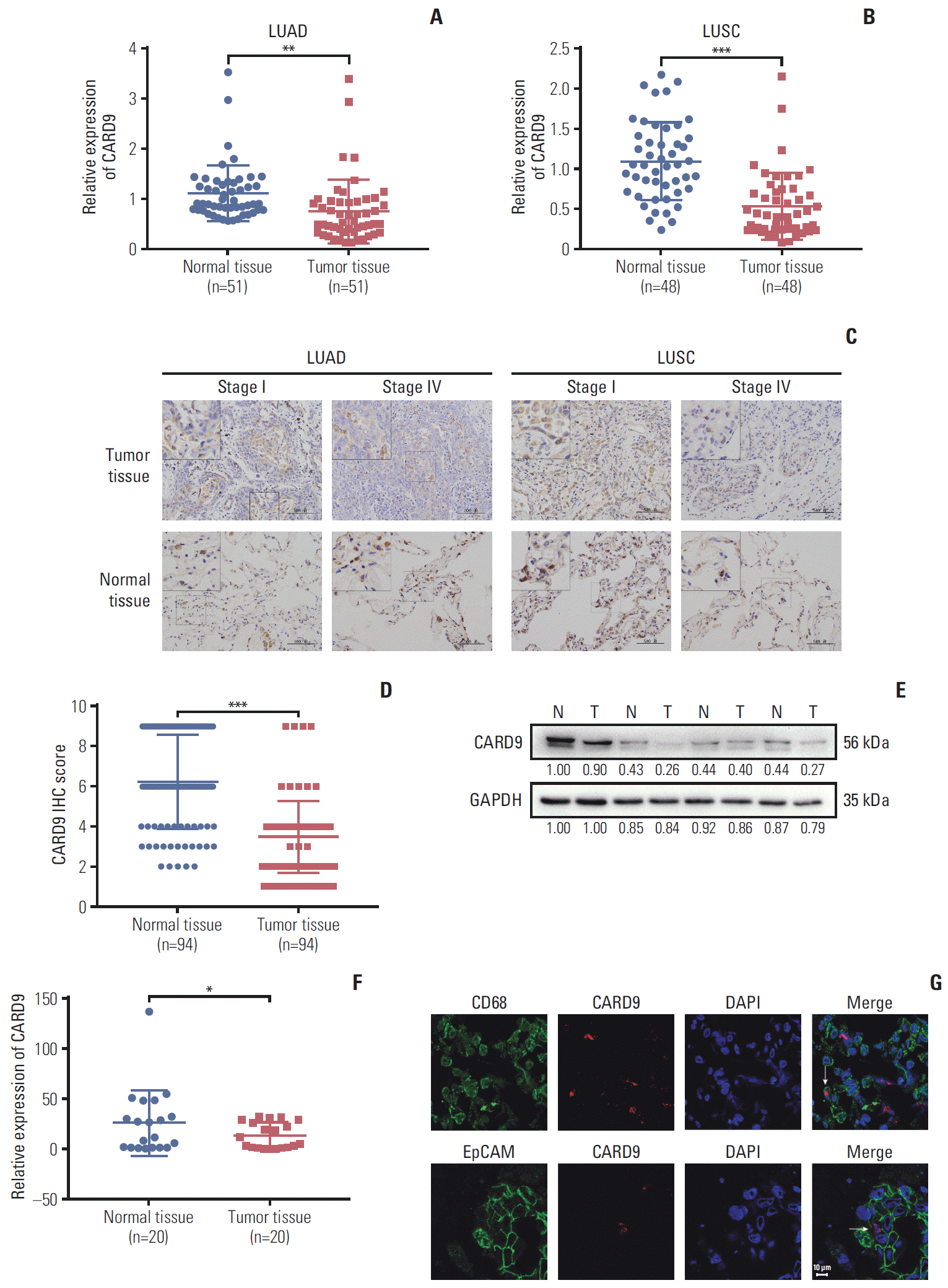
Fig. 1.
Caspase recruitment domain containing protein 9 (CARD9) was decreased at the level of mRNA and protein in non-small cell lung cancer (NSCLC) tissues. (A, B) CARD9 mRNA level between tumor tissues and normal tissue in lung adenocarcinoma and lung squamous cell carcinoma based on The Cancer Genome Atlas database lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) projects. (C) Expression of CARD9 in LUAD and LUSC tissues and paired adjacent normal lung tissues by immunohistochemistry. (D) Comparison of immunohistochemistry score of CARD9 expression in 94 NSCLC tissues and paired adjacent normal lung tissues. tissues. (E) CARD9 expression in tumor tissues (T) and normal lung tissues (N) by western blot. (F) CARD9 mRNA level of 20 pairs of tumor tissues and normal lung tissues by real-time quantitative reverse transcription polymerase chain reaction. (G) Immunofluorescence scanning of tumor tissues from NSCLC patient. Anti-CD68 was for staining macrophages and anti–epithelial cell adhesion molecule (EpCAM) for tumor cells. White arrows showed the CARD9-expressing macrophages and tumor cells. Scale bar=10 μm. Values are presented as mean±standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001.
![]()
2. CARD9 downregulation was associated with poor prognosis in NSCLC patients
Based on TCGA datasets, the level of CARD9 in LUAD was negatively associated with TNM stage, tumor size, lymph node metastasis, and remote metastasis (Table 1). Consistent with this, patients with higher expression of CARD9 in LUAD had better OS than patients with lower expression of CARD9 (hazard ratio [HR], 0.710; p=0.023) (Fig. 2A). However, CARD9 expression in LUSC was only related to lymph node metastasis (Table 2), and OS in LUSC patients was not significantly different regardless of CARD9 expression (HR, 1.233; p=0.152) (Fig. 2B). We surmised that CARD9 had higher clinical value for LUAD. As shown in Table 3, CARD9 was negatively correlated with TNM stage (p < 0.001), histological differentiation (p=0.002), T category (p=0.001), lymph node metastasis (p=0.040), and remote metastasis (p < 0.001). The IHC results also showed decreased CARD9 in Ⅳ stages of NSCLC compared with Ⅰ stages (Fig. 1C). And the group with higher CARD9 expression had better OS (HR, 0.304; p=0.027) (Fig. 2C) and PFS (HR, 0.310; p=0.004) (Fig. 2D) than the group with lower CARD9 expression. We analyzed the relationship between CARD9 and prognosis in patients of LUAD in our cohort. Similarly, patients with lower CARD9 expression had a worse OS (HR, 0.161; p=0.009) (Fig. 2E) and PFS (HR, 0.326; p=0.019) (Fig. 2F). Logistic regression analysis showed that the high expression of CARD9 was an independent protective factor for OS (odds ratio [OR], 0.127; p=0.022) and PFS (OR, 0.126; p=0.004), excluding confounding factors of age, sex, smocking status, and TNM stage. Therefore, we speculated that CARD9 might be a potential predictor of prognosis for NSCLC patients.
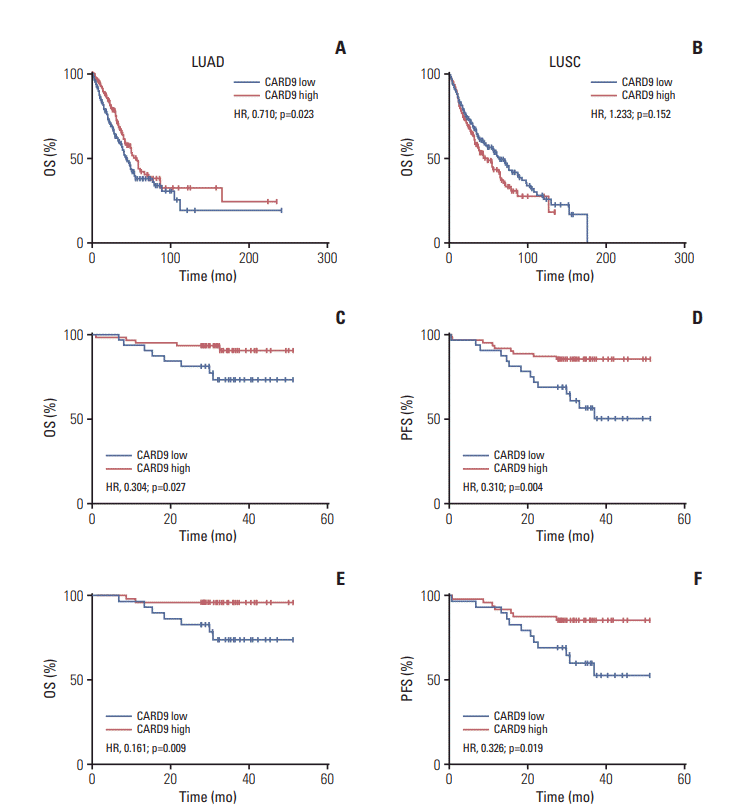
Fig. 2.
Decreased expression of caspase recruitment domain containing protein 9 (CARD9) correlated with poor prognosis. (A, B) Overall survival (OS) of lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) from The Cancer Genome Atlas database. (C, D) The OS and progression-free survival (PFS) of our clinical cohort of non-small cell lung cancer patients. (E, F) The OS and PFS of our clinical cohort of LUAD patients.
![]()
Table 1.
Correlation between CARD9 expression and the clinicopathological features of LUAD patients from TCGA database
![]()
Table 2.
Correlation between CARD9 expression and the clinicopathological features of LUSC patients from TCGA database
![]()
Table 3.
Correlation between CARD9 expression and the clinicopathological features of NSCLC patients (n=94)
![]()
3. CARD9 downregulation promoted NSCLC cell migration and invasion
Since CARD9 was expressed both in tumor cells and macrophages, we analyzed the baseline expression of CARD9 in NSCLC cell lines (including H1299, A549, 95C, 95D, and PC9), HBE, human normal lung epithelial cells (BEAS-2B), and human leukemic monocyte (THP-1). Because of the high CARD9 production in THP-1, we loaded less concentration of THP-1 protein for easier observation of CARD9 in other cells. Under this circumstance, PC9, H1299, and HBE had detectable CARD9 expression at protein level, among which PC9 had the highest CARD9 expression (Fig. 3A). PCR result showed that, unlike western blot to some extent, 95D cells had comparable CARD9 mRNA to HBE and H1299 cells, which may be due to the regulation of posttranscriptional process (Fig. 3B). Considering CARD9 was also expressed in tumor cells and varied in different NSCLC cell lines, which indicated its potential participation in tumorigenesis, we first focused on the study of CARD9 in tumor cells. We generated H1299 and PC9 cells stably expressed CARD9 or control vector. On the other hand, CARD9-target shRNA successfully knocked down CARD9 in PC9 cells and shControl was served as control (Fig. 3C and D). Since H1299 was relatively scarce in CARD9 production compared with THP-1 and PC9, H1299 shCARD9 was not generated. Wounding healing showed the migratory ability of H1299 and PC9 was inhibited by ectopic CARD9 (p=0.002 and p=0.007, respectively) (Fig. 3E) while decreased CARD9 markedly promoted PC9 migration (p=0.030) (Fig. 3E). Meanwhile, in the matrigel invasion assays, CARD9 was found to block invasiveness capability of H1299 and PC9 (p=0.002 and p=0.010, respectively) (Fig. 3F), and when its expression was suppressed in PC9, invasion was significantly augmented (p=0.040) (Fig. 3F). We also tested epithelial-mesenchymal transition (EMT)–associated protein. In consistent with these results, increased CARD9 was accompanied with decreased N-cadherin and transforming growth factor-β, which were able to facilitate EMT process [12], though E-cadherin and vimentin were unch-anged (Fig. 3C). These suggested that CARD9 acted as an inhibitory factor in cancer progression through weakening tumor migration and invasion ability.
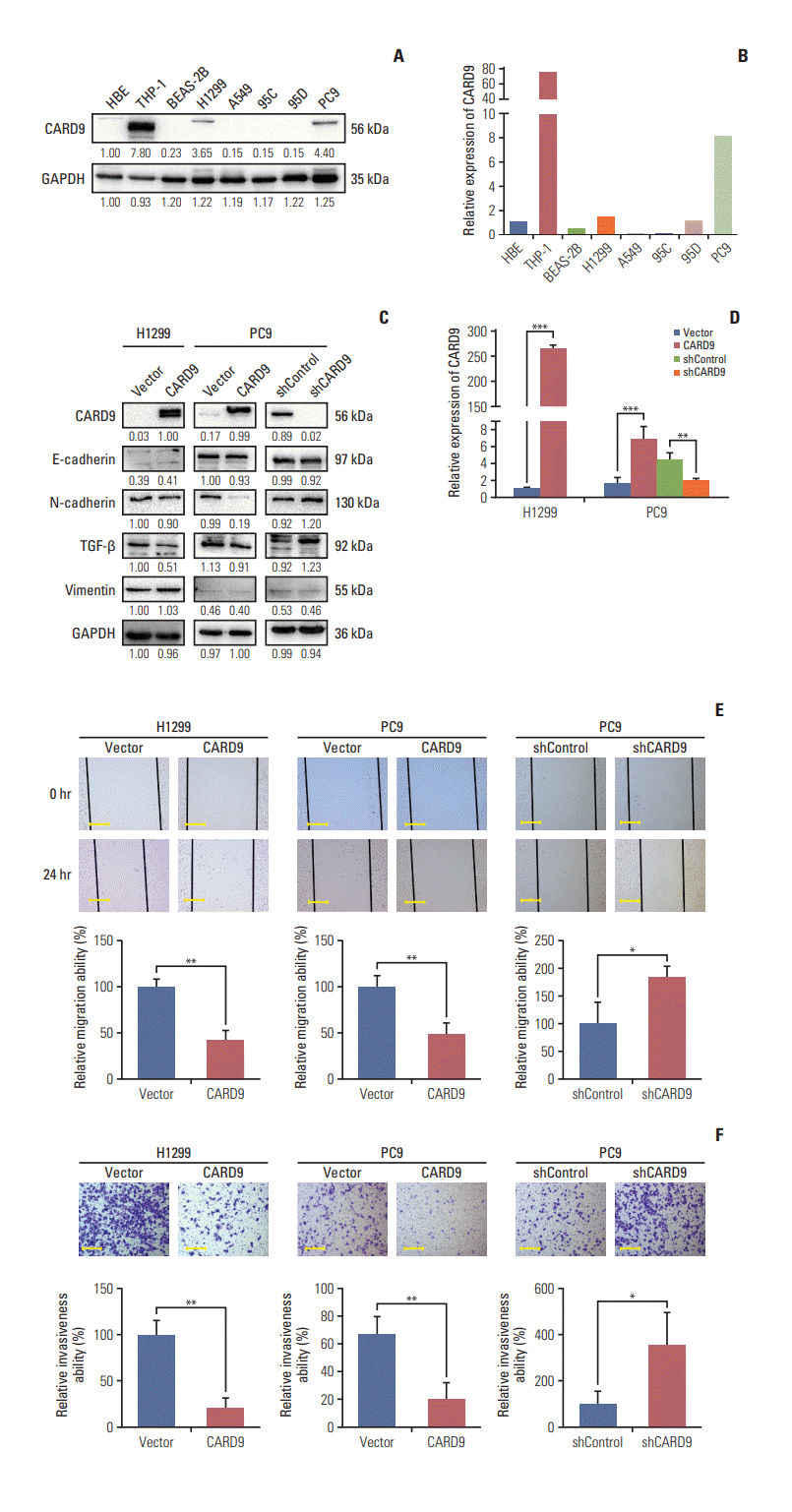
Fig. 3.
Downregulated caspase recruitment domain containing protein 9 (CARD9) promoted non-small cell lung cancer (NSCLC) migration and invasion. (A, B) Baseline of CARD9 in human bronchial epithelioid cells (HBE), human leukemic monocyte (THP-1), BEAS-2B cells (human normal lung epithelial cells), and NSCLC cell lines (H1299, A549, 95C, 95D, and PC9) by western blot. (C) Western blot of CARD9, E-cadherin, N-cadherin, transforming growth factor β (TGF-β), and vimentin in CARD9-overexpressing H1299 and PC9, CARD9-knockdown PC9, and their control cells. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the internal reference. Numbers under the bank presented relative expression of protein evaluated by Image J. (D) Reverse transcription polymerase chain reaction was used to determine successful cells transfection. (E, F) Migration and invasion abilities of CARD9-overexpressing H1299 and PC9 cells, and CARD9-knockdown PC9 cells compared with their control cells. Scale bar=50 μm. Experiments were performed in triplicate and values are represented as mean±standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001.
![]()
4. CARD9 inhibited NSCLC cells proliferation and promoted apoptosis in vitro
Then we investigated the effect of CARD9 on cell proliferation and apoptosis. Cell growth curve assays showed CARD9-overexpressing H1299 and PC9 had suppressed growth, and CARD9-knockdown PC9 had promoted growth (p < 0.001) (Fig. 4A). Consistently, cell clonal formation assays showed dampened clonal formation in high-CARD9–expressing H1299 and PC9 cells compared with their control counterparts (p=0.036 and p=0.027, respectively) (Fig. 4B), while CARD9-knowdown PC9 cells had elevated clonal numbers (p < 0.001) (Fig. 4B). We further assessed the effect of CARD9 on cell cycle. Ectopic expression of CARD9 in H1299 significantly induced G0/G1 arrest, exhibiting increased G0 to G1 phase cells (p=0.008) (Fig. 4C) and decreased S-phase population (p=0.004) (Fig. 4C). In PC9 cells, overexpression of CARD9 also resulted in increased G0 to G1 phase cells (p=0.001) (Fig. 4C) with a corresponding decrease in G2-phase population (p=0.016) (Fig. 4C). On the other hand, knockdown of CARD9 in PC9 cells presented decreased population of G0 to G1-phase (p < 0.001) (Fig. 4C) with elevated population of S- and G2-phase (p=0.001 and p < 0.001, respectively) (Fig. 4C), suggesting that downregulation of CARD9 would drive the transition from G1 to S. Consistent with this, CyclinD1, necessitated for cell cycle progression, was significantly attenuated when CARD9 was overexpressed in H1299 and PC9 cells and was augmented when CARD9 was decreased in PC9 cells (Fig. 4E). In addition, by examining apoptosis by flow cytometry, we found that CARD9 could lead to more late apoptosis in H1299 and PC9 cells (p < 0.001 and p < 0.001, respectively) (Fig. 4D). However, once it was knockdown, population of late apoptosis was consistently reduced (p < 0.001) (Fig. 4D). The early apoptosis is not affected. This supportive role of CARD9 on apoptosis was further evidenced by higher expression of cleaved-caspase-3 accompanied with higher level of CARD9 (Fig. 4E). Taken together, CARD9 could impede NSCLC cell proliferation but enhance apoptosis in vitro.
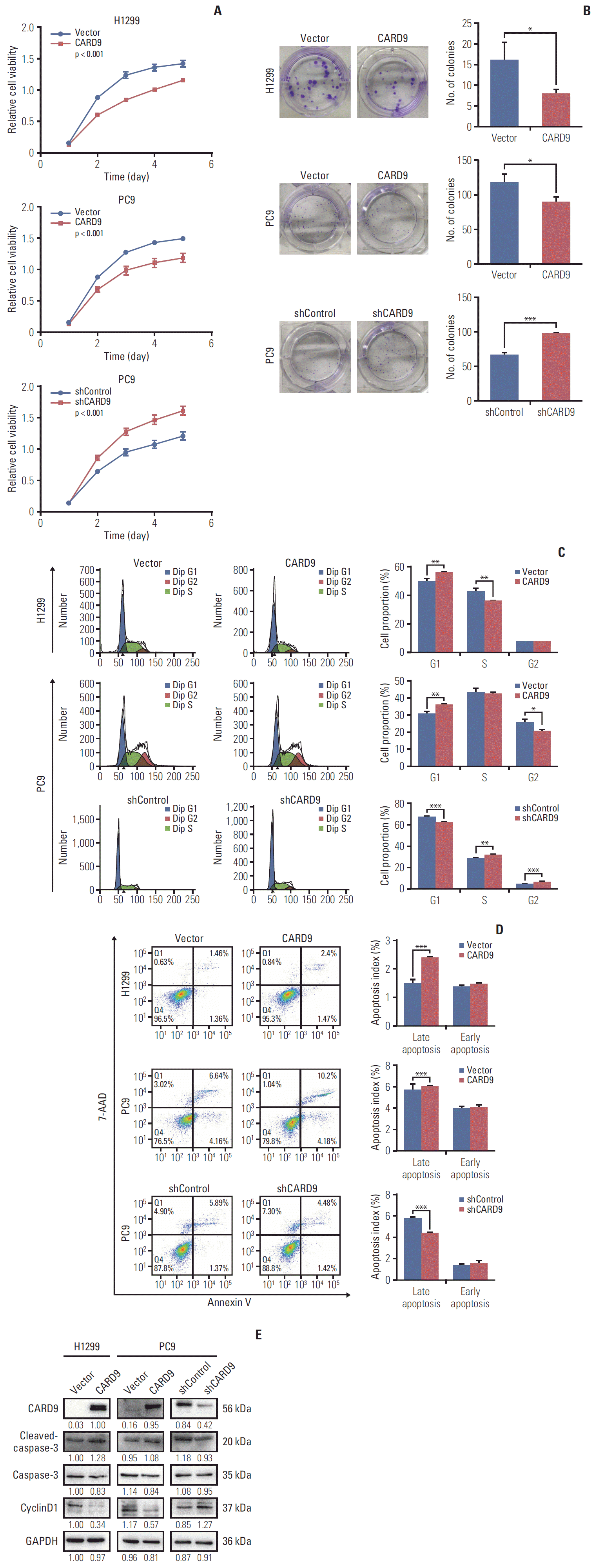
Fig. 4.
Caspase recruitment domain containing protein 9 (CARD9) suppressed cells proliferation but supported cell apoptosis. (A, B) Cell growth assay and clonal formation assay of CARD9-overexpressing H1299 and PC9 cells, PC9 shCARD9, and their control cells. (C, D) Analysis of cell circle progression and apoptosis of CARD9-overexpressing H1299 and PC9 cells, CARD9-knockdown PC9 and their control cells. (E) Western blot of CARD9, cleaved-caspase-3, caspase-3, and cyclin D1. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the internal reference. Numbers under the bank presenting relative expression of protein evaluated by Image J. Experiments were performed in triplicate and values are represented as mean±standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001.
![]()
5. CARD9 interacted with BCL10 and inhibited MAPK/p38 signaling
Previous studies had found that in anti-infection immunity, CARD9 recruited BCL10 and mucosa-associated lymphoid tissue 1 (MALT1) to form CBM (CARD9-BCL10-MALT1) complex [13,14], activating downstream such as nuclear factor κB (NF-κB) and MAPK signaling [3,6,13,15]. Surprisingly, in our study, we found that MAPK/p38 was inactivated when CARD9 was upregulated and the phosphorylation of p38 was facilitated when CARD9 was inhibited (Fig. 5A). But the other two subunits of MAPK, extracellular signal-related kinase, and JNK (c-Jun N-terminal kinase), exhibited no significant change (Fig. 5A). As for NF-κB signaling, phosphorylation of p50 and p65 were declined in CARD9-overexpressing H1299 and PC9 cells but remained unaltered in CARD9-knockdown PC9 cells (Fig. 5B). To testify whether BCL10 interacted with CARD9, we performed Co-IP in PC9 shControl, PC9 shCARD9, H1299 Vector, and CARD9-overexpressing H1299 cells. As shown in Fig 5C, in PC9 shControl cells, BCL10 was found to bind to CARD9, while in PC9 shCARD9 cells, less CARD9 was pulled down and interacting BCL10 was not detected. In H1299 cells, CARD9 was still undetectable by using CARD9 antibody in H1299 Vector cells, but when CARD9 was overexpressed, BCL10 was found in the elute lysates. These results demonstrated that BCL10 interacted with CARD9. We also searched String database [11] for well-established network proteins of CARD9. It showed that BCL10 was the known protein interacting with CARD9, and in keeping with our results, there were three p38 MAPKs (MAPK11, MAPK12, and MAPK14) associated with CARD9 (Fig. 5D).
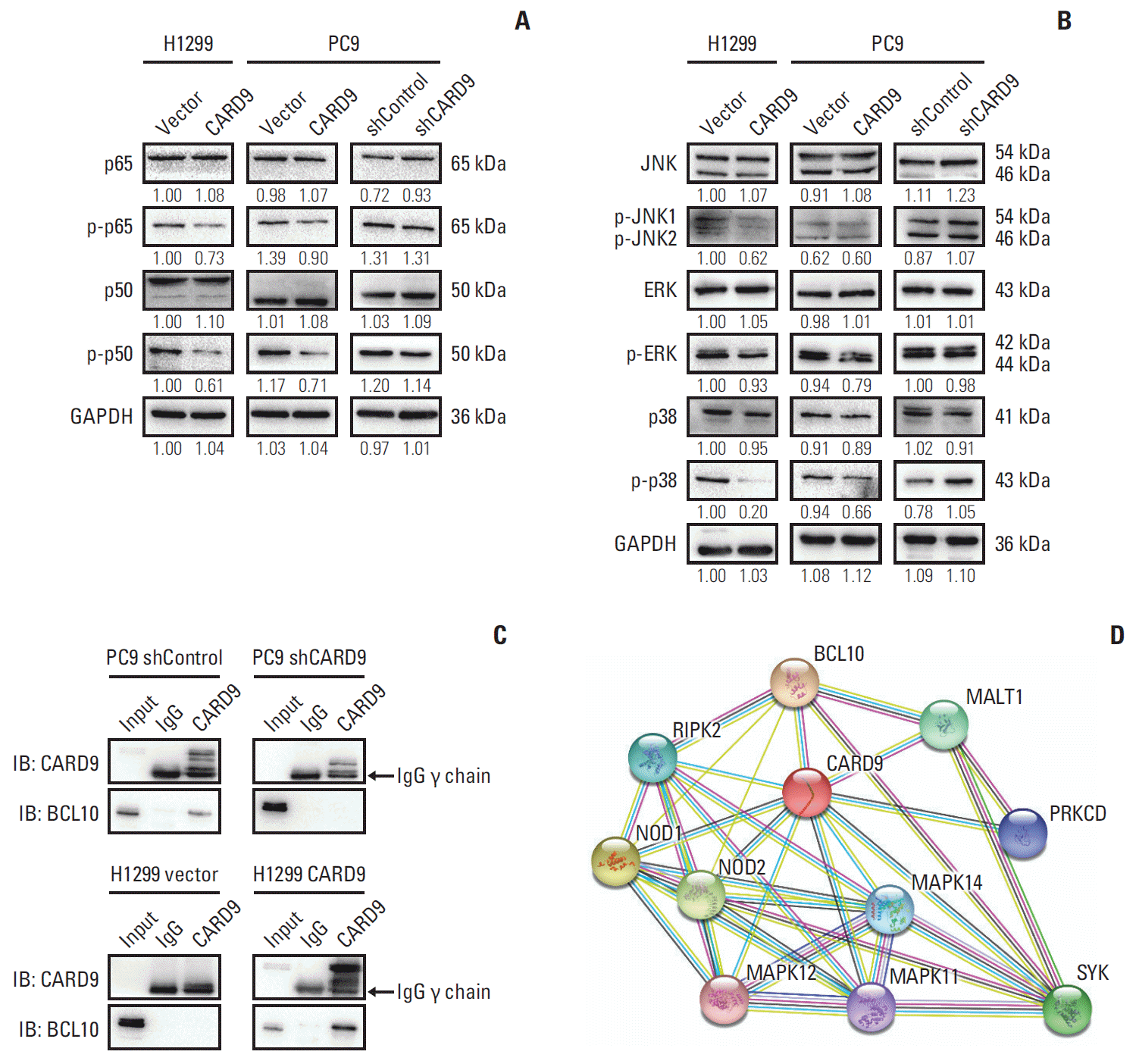
Fig. 5.
Caspase recruitment domain containing protein 9 (CARD9) interacted with BCL10 and inhibited p-p38 activation. (A, B) Western blot of nuclear factor κB signaling and mitogen-activated protein kinases (MAPK) signaling in CARD9-overexpressing H1299 and PC9 cells, CARD9-knockdown PC9 and their control cells. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the internal reference. Numbers under the bank presenting relative expression of protein evaluated by Image J. (C) The interaction between CARD9 and BCL10 was confirmed by coimmunoprecipitation in PC9 cells and H1299 cells. IB, immunoblot. (D) Interacting protein network of CARD9 was searched from String database.
![]()
6. CARD9 suppressed cell proliferation, migration, and invasion via inhibition of MAPK/p38 signaling
Considering the consistent change of p-p38 in our established cell lines, we next explored which cancerous capacities were regulated by CARD9 in a p-p38-dependent manner. We used a p38-kinase–specific inhibitor SB203580 to inhibit phosphorylation of p38 in CARD9-knockdown PC9 cells and DMSO served as control (Fig. 6A). The cell growth curve assays showed a dramatical impeded growth in SB203580-treated PC9 shCARD9 cells (p < 0.001) (Fig. 6B) while the growth of PC9 shControl cells was unaffected by SB203580 (Fig. 6B). The cell clonal formation assays showed no significant difference between PC9 cells and their SB203580-treated counterparts (Fig. 6C). In addition, retarded activation of p38 significantly limited the invasiveness of PC9 shControl and PC9 shCARD9 cells (p=0.009 and p=0.039, respectively) (Fig. 6D). Migration ability was also limited both in shControl and shCARD9 PC9 cells by SB203580 (p=0.021 and p=0.002, respectively) (Fig. 6E). We further explored whether cell apoptosis and cycle progression were controlled by p38 signaling. Of note, late apoptosis in shControl PC9 cannot be induced but even be hindered by SB203580 (p=0.001) (Fig. 6F), though both late and early apoptosis of shCARD9 PC9 cells was indeed increased when intervened with SB203580 (p=0.046 and p=0.013, respectively) (Fig. 6F). This phenomenon also happened in cell cycle progression analysis. We found shCARD9 PC9 cells had accumulated G0 to G1 phase population (p=0.001) (Fig. 6G) and declined S-phase population (p=0.001) (Fig. 6G) when p-p38 was blocked, but shControl PC9 group instead had slightly increased S-phase population with SB203580 treatment (p=0.020) (Fig. 6G). Studies have shown that abrogation of overactivated p38 signaling was able to reverse enhanced G1 to S transition and anti-apoptosis effect in CARD9-knockdown cells but not in normal PC9 cells. This contradictory result may be due to activation of bypass signal when low level of p-p38 was suppressed again in shControl PC9 cells. Taken together, CARD9 impaired abilities of cell proliferation, migration, and invasion by suppressing MAPK/p38 signaling.
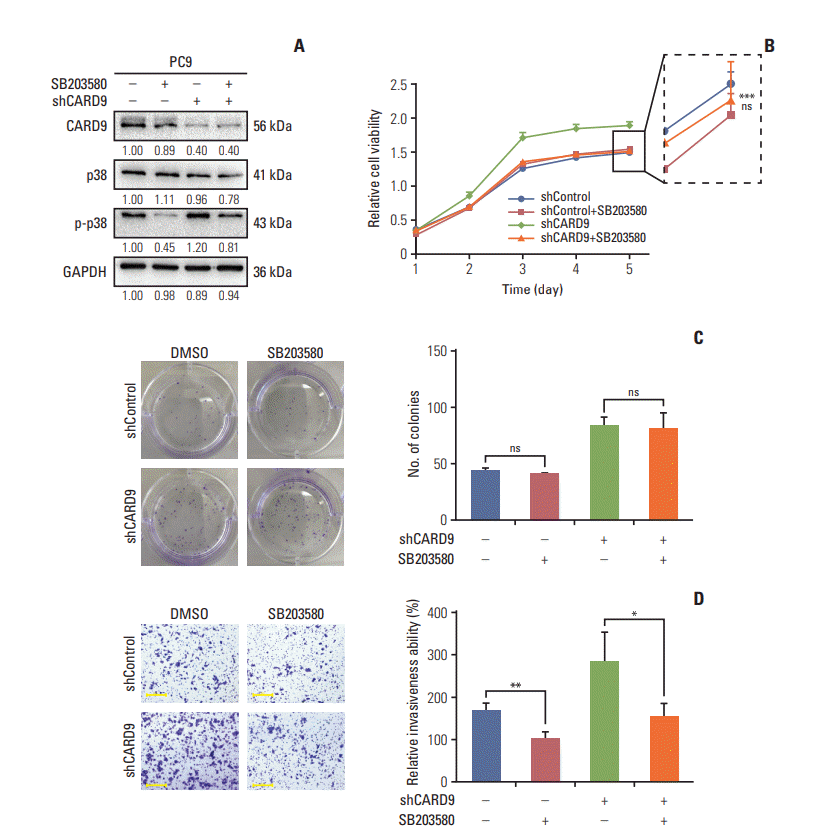
Fig. 6.
Caspase recruitment domain containing protein 9 (CARD9) suppressed non-small cell lung cancer proliferation and promoted apoptosis via inhibition of mitogen-activated protein kinase/p38 activation. (A) Western blot of p-p38 in PC9 shCARD9 and PC9 shControl cells. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the internal reference. Numbers under the bank presenting relative expression of protein evaluated by Image J. (B, C) Cell growth assay and clonal formation assay of SB203580-treated PC9 shCARD9 and PC9 shControl cells and their counterparts. ***The comparison between SB203580-treated PC9 shCARD9 and PC9 shCARD9 (p < 0.001); ns, the comparison between SB203580-treated PC9 shControl and PC9 shControl. DMSO, dimethyl sulfoxide. (D, E) Invasion and migration abilities of SB203580-treated PC9 shCARD9 and PC9 shControl cells and their counterparts. Scale bars=50 μm. (F, G) Analysis of apoptosis and cell circle progression in SB203580-treated PC9 shCARD9 and PC9 shControl cells and their counterparts. Experiments were performed in triplicate and values are represented as mean±standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001.
![]()
Discussion
Research on the role of CARD9 in malignant diseases, especially the role of CARD9-mediated immunity or inflammation in tumors has been a hotspot nowadays. Contrary to previous findings regarding the increase of CARD9 in tumors, our previous research found that CARD9 was decreased in malignant pleural effusion compared with benign pleural effusion, indicating an interesting role of CARD9 in lung cancer [9]. In this study, both TCGA database and our clinical cohort showed the mRNA and protein level of CARD9 were lower in NSCLC tissues than that in adjacent normal lung tissues. Lower CARD9 expression was related to worse OS and PFS of NSCLC patients, and CARD9 was an independent prognostic factor. In vitro experiments showed that upregulated CARD9 could inhibit NSCLC cells growth, migration, invasion and induce cell apoptosis via suppression on p38 signaling. Therefore, we inferred that CARD9 may play an anti-tumor role in lung tumor progression and metastasis which could serve as a potential target for diagnosis and treatment.
However, most studies about the effect of CARD9 on cancer focused on myeloid cells, especially macrophages in tumor microenvironment (TME), and their results were still incongruent. In colon cancer, CARD9 in macrophages can promote tumor invasion and metastasis by driving bias of tumor-associated macrophages towards M2-macrophages [16]. Bergmann et al. [17] also revealed that in colitis-associated cancer (CAC), CARD9 was indispensable to interleukin1β (IL-1β) production from myeloid cells, which promoted tumor progression. However, other studies held the opposite view. It was indicated that Dectin-1 activation could reverse M2 phenotype in mice models bearing lung cancer, mammary carcinoma, and melanoma [18], and enhance T-cell cytotoxicity in melanoma mice model [19], both in a CARD9-dependent way. Qu et al. [10] discovered that CARD9 in MDSCs inhibited lung cancer development, in line with our result that CARD9 was an antitumor factor for lung cancer.
In our study, both tumor cells and macrophages were found to produce CARD9. In our opinion, for one thing, the expression of CARD9 varied in different NSCLC cells, a suggestion of important role of CARD9 within NSCLC cells for tumor development. For another, it is typical of tumor to accumulate myeloid cells [10], such as macrophages [12,20], which should lead to increased CARD9 in vivo. We surmised that dramatical downregulation of CARD9 in NSCLC cells contributed to the decreased CARD9 in vivo, which may play a role in NSCLC development. And CARD9 in renal carcinoma cells had been substantiated to induce EMT by stimulating JNK/AP1 signaling [4,21]. Consequently, we started on our first step in studying the role of CARD9 in lung tumor cells.
Previous studies have demonstrated that CARD9 transmitted signals from Dectin-1 [22] to activate p38 pathway for pro-inflammatory cytokines in anti-fungal immunity, and similarly, NOD2 (nucleotide-binding oligomerization domain containing)-CARD9-p38 axis was activated for anti-bacterial immunity [3]. But in present study, activation of p38 was blocked by CARD9 to weaken malignant biological characteristics of NSCLC cells. This negative regulatory ability of CARD9 had been reported [3,23]. Hsu et al. [3] found that the CARD itself could inhibit NOD2-induced p38 activation; Pereira et al. [23] demonstrated that NOD2-CARD9 axis negatively regulated synthesis of pro–IL-1β via suppression on MAPK signaling in Salmonella infection. Therefore, CARD9 as an adaptor in innate immunity, its effect on p38 was dependent on the stimulating signals upstream. But how CARD9 in tumor cells regulated p38 had not been elucidated yet. Based on great potential of p38-targeted therapy in liver cancer treatment [24,25], downregulated-CARD9–induced p38 activation was an important implication for clinical application of p38-targeted therapy in NSCLCs.
Tumor initiation and development are affected by internal gene factor, and external factors such as inflammation, TME, and even infection. Recently, two new studies together explained CARD9 could also influence tumorigenesis via fungal-associated way. Malik et al. [26] and Wang et al. [27] both found that CARD9–/– mice was more susceptible to chemical-induced colon cancer with changed fungal constitution in colon, while the underlying mechanisms were narrated differently. On the one hand, when infected by fungus, deficient CARD9 could cause a blocked activation of inflammation and production of IL-18, which was critical for epithelial restitution and T cells response [26]. On the other hand, uncontrolled fungal burden led to overwhelming inflammatory cytokines, such as IL-6 and granulocytemacrophage colony stimulating factor, which promoted upregulation of MDSCs and then downregulation of cytotoxic T cells [27]. Both roads were to the development of CAC. Based on the CARD9-associated anti-infection in colon carcinogenesis, lung as another microbial-enriched organ, CARD9 may also have an unknown role in lung tumor initiation.
Above all, the complexity of innate immunity determined the multifaced roles of immune-cell–derived CARD9. CARD9 in immune cells could influence tumor initiation and development from fungal, inflammation, and TME perspectives. Our exploration into CARD9 of macrophages in NSCLC development is on the schedule. CARD9 in tumor cells also had effects on tumor development. Hence, manipulating function of CARD9 within certain cells is critical for elaborating roles of CARD9 and investigating therapy targeting on CARD9. Our research chose the NSCLC cells as our first step to explore its potential for prognosis predictor and targeted therapy, which will be followed more in-depth and multi-directional studies in the future.
 XML Download
XML Download