

 PDF
PDF Citation
Citation Print
Print


Introduction
Hereditary cancer syndrome means that inherited genetic mutations can increase a person’s risk of developing cancer. Specifically, certain genetic mutations can cause changes in the growth control of normal cells and cause them to become cancerous. Genetic mutations that promote cancer can be inherited if the mutations are present in germ cells. It is reported that inherited genetic mutations play a major role in 5% to 10% of all cancers. The most well-known genes associated with hereditary cancer syndrome are the BRCA1/2 genes for hereditary breast and ovarian cancer syndrome (HBOC) and the TP53 gene for Li-Fraumeni syndrome. Approximately 7% of breast and 13% of ovarian cancers are estimated to be due primarily to germline mutations in the BRCA1/2 genes. The cumulative risks of breast and ovarian cancers in BRCA1/2 mutation carriers are reported to be 72% (95% confidence interval [CI], 65 to 79) and 44% (95% CI, 36 to 53), respectively, in BRCA1 carriers and 69% (95% CI, 61 to 77) and 17% (95% CI, 11 to 25), respectively, in BRCA2 carriers [1]. The cumulative cancer risk associated with TP53 mutation may be as high as 90% by the age of 60 years [2]. In addition to mutations in BRCA1/2 and TP53, germline mutations in certain genes were associated with more than 50 hereditary cancer syndromes. Genetic tests for hereditary cancer syndromes can identify individuals and families at increased risk of developing cancer. Once individuals or families are identified for hereditary cancer syndrome, they can be referred for risk assessment and personalized management that may include intensive cancer surveillance, risk-reducing surgery and genetic counseling.
With the rapid progress that has been made in next-generation sequencing (NGS) technology, simultaneous sequencing of multiple genes has become available through multiple-gene panel testing, which is less expensive and more rapid than single-gene testing. Furthermore, multiple-gene panels using NGS technology have increased the detection rate of mutations compared to conventional gene-bygene testing [3].
Currently, several commercial multiple-gene panels provide genetic information for hereditary cancer risk assessment. However, there are differences among ethnicities in cancer-susceptible germline mutations, and the assessment of germline mutations in all ethnic groups with clinical data is mandatory. In Korea and Asia, several studies evaluated the frequency of germline mutations, including BRCA1/2 and/or other mutations associated with hereditary cancer syndrome. However, the results of most of the studies were not representative of the Korean and Asian population because of the relatively small number of patients included and the limited gene list evaluated.
In this study, we applied multiple-gene panel testing to 64 cancer susceptibility genes to examine the frequency of mutations and to assess the clinical value of NGS-based multiple-gene panel testing in breast cancer patients with clinical features of HBOC.
Materials and Methods
1. Patient selection
The study population included breast cancer patients with the following features of HBOC: (1) diagnosed with breast cancer and another primary cancer; (2) a family history that included at least two cases of breast cancer in first- or second-degree relatives; (3) bilateral breast cancer; or (4) breast cancer diagnosis before the age of 40 years and with collected blood samples in tissue bank in Breast Care Center, Seoul National University Hospital, Korea and Breast Cancer Center, National Cancer Center, Korea (CONSORT diagram) (S1 Fig.) Of the patients, 349 patients were admitted to Seoul National University Hospital, Korea, and 147 patients were admitted to the National Cancer Center, Korea, between 2002 and 2017. All patients consented to multi-gene panel testing for clinical research. Blood samples of the included patients were collected from each hospital and sent to a central laboratory for sequencing. The medical records were reviewed, and personal and family histories and pathologic data of cancer were recorded.
2. NGS assay
Genomic DNA was extracted from the participants’ peripheral blood samples. Our panel included 64 hereditary cancer-predisposing genes (ALK, APC, ATM, ATR, BAP1, BARD1, BLM, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, FAM175A, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FH, FLCN, GSTP1, HOXB13, KRAS, LIG4, MEN1, MET, MLH1, MRE11A, MSH2, MSH6, MUTYH, NAT1, NBN, NF1, PALB2, PALLD, PMS2, PRKAR1A, PRSS1, PTEN, RAD50, RAD51, RAD51C, RAD51D, RB1, RET, SDHB, SDHC, SDHD, SLX4, SMAD4, SPINK1, STK11, TP53, VHL, and XRCC2) (Table 1). For mutation analysis, 64 gene-containing DNA fragments were enriched by solution-based hybridization capture and followed by sequencing with an Illumina NextSeq platform (Illumina, San Diego, CA) with the 150-bp paired-end read module. The target region included all coding exons. Capture probes were generated by Celemics, Inc. (Seoul, Korea). The hybridization capture procedure was also performed according to the manufacturer’s standard protocol. Genomic DNA was sheared via sonication. Biotynilated RNA oligonucleotide probes were hybridized with sheared DNA. Captured fragments were removed from solution via streptavidin-coated magnetic beads and subsequently eluted. The enriched fragment library was then subjected to polymerase chain reaction (PCR) amplification using primers specific to the linked Illumina adaptors. Resulting libraries were quantified via Agilent 2200 TapeStation before proceeding to Illumina NextSeq platform. All samples were pooled into a single lane on a flow cell and sequenced together.
Raw FASTQ files were filtered using Trimmomatic ver. 0.33 and aligned with the genome of reference (GRCh37/hg19) using Burrows-Wheeler Aligner ver. 0.7.10. PCR duplicates, overrepresented sequences, and low-quality reads were removed. Realignments of insertions and deletions were performed using GATK. Reads with mapping quality of 0 were filtered out. If a read was able to be mapped at two different places with an identical percentage, the mapping quality equaled zero. Otherwise, the read was mapped to the most identical region. When ≥ 90% of mutation reads were biased to forward or reverse, this read was filtered out. The cutoff value of minimum supporting reads and minimum coverage was 2 and 8. The mean depth over target region was 660. The mean read size was 139 and mean uncovered ratio over target was 0.16%. Variant calling was performed with Samtools ver. 1.1 and Varscan ver. 2.4.0 (S2 Table).
3. Mutation analysis and variant classification
Variants were described according to the nomenclature recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen) and classified according to the following American College of Medical Genetics and Genomics recommendations: pathogenic (P), likely-pathogenic (LP), variants of unknown significance (VUS), likelybenign, and benign/polymorphism [4]. We used online databases, including the Human Gene Mutation Database, the Single Nucleotide Polymorphism Database, the 1000 Genome project, ClinVar, the Sorting Intolerant From Tolerant, Polymorphism Phenotyping-2, and the Korean Reference Genome Database, for in silico prediction of identified variants. Variants classified as P or LP were considered deleterious mutations.
4. Statistical analysis
Participant characteristics and sequencing results were summarized with descriptive statistics, which included medians, means, and standard deviations. The distributions of deleterious mutation according to the inclusion criteria were compared using Pearson’s chi-square analysis and Student’s t test. All p-values were 2-sided and a p-value less than 0.05 was considered significant. All statistical analyses were performed using IBM SPSS Statistics for Windows ver. 23.0 (IBM Corp., Armonk, NY).
Results
1. Study population
The clinical characteristics of the patients are shown in Table 2. The median age at diagnosis of cancer was 48 years (range, 19 to 80 years). In these patients, 390 patients (78.6%) had stage I or II disease. More than half of the patients (n=250, 50.4%) had another primary cancer, including ovarian cancer, stomach cancer, colon cancer, lung cancer, or other malignancy. In all, 169 patients (34.1%) reported that they had two or more first- or second-degree relatives with breast cancer. Fifty-seven patients (11.5%) had synchronous or metachronous bilateral breast cancer, and 84 patients (16.9%) were diagnosed with breast cancer at an age younger than 40 years. Sixty-four patients had two or more risk factors for HBOC (e.g., bilateral breast cancer and breast cancer diagnosis < 40 years old).
2. Frequency of deleterious mutations
Table 2 summarizes the characteristics of patients with and without deleterious mutations. Of all 496 patients, 95 patients (19.2%) were found to have deleterious germline mutations of cancer susceptibility genes and 401 patients (80.8%) were not detected to carry deleterious mutations. The breast cancer stage was not different between the two groups (p=0.078). The proportions of risk factors, including breast cancer with another primary cancer, family history of breast cancer, and bilateral breast cancer were also not different between the groups. However, the proportion of patients with deleterious mutations were higher in patients who were diagnosed with breast cancer at younger than 40 years old than patients with another risk factors. Breast cancer diagnosis at young age was associated with a higher rate of deleterious mutations (p=0.022). Furthermore, having two or more risk factors for HBOC was also associated with a higher rate of deleterious mutations (p=0.001).
Breast cancers can be divided into four major subtypes depending on hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) status that have different clinical outcomes and responses to therapy a: luminal A (HR+ and HER2–), luminal B (HR+ and HER2+), HER2-enriched (HR– and HER2+), and triple-negative (HR– and HER2). Table 3 showed the percentage of subtypes in 410 patients whose immunohistochemistry data were available. The percentage of patients with BRCA1 mutations was different with patients without BRCA1/2 mutations (p < 0.001). Seventy-five percent of patients with BRCA1 mutations were triple-negative breast cancer, whereas 20.8% of patients were HR (+) breast cancer including luminal A and luminal B subtypes. In contrast, the percentage of subtypes in BRCA2 mutations was not statistically different with patients without BRCA1/2 mutations (p=0.825).
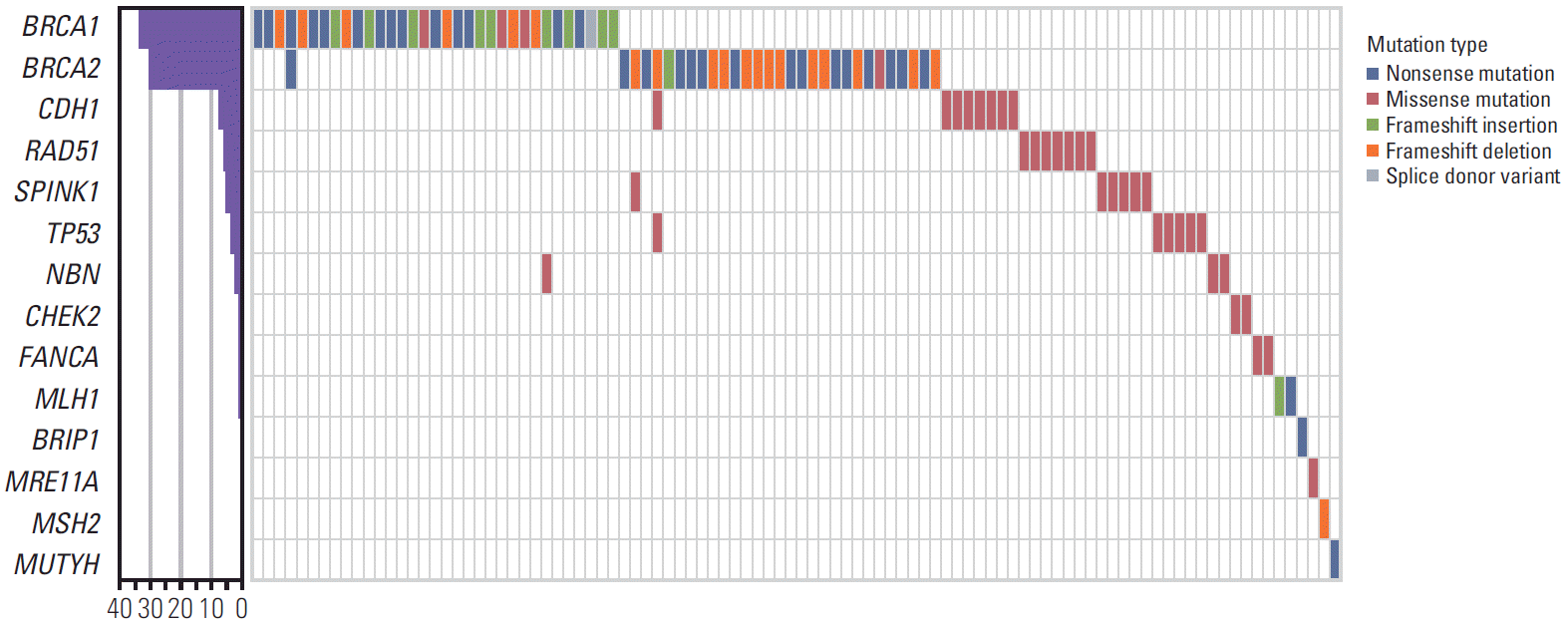
Table 4 and Fig. 1 summarize 48 deleterious mutations found in 95 patients. Of these patients with deleterious mutations, 60 patients (12.1%) had BRCA1/2 mutations: 31 in BRCA1 and 30 in BRCA2. Patients HOPE_309 and HOPE_502 had 2 BRCA1 mutations and patient HOPE_57 carried both BRCA1 and BRCA2 mutations. In addition, 38 patients (7.7%) had cancer susceptibility gene mutations other than BRCA1/2: 35 patients had non-BRCA1/2 mutations and three patients had both a BRCA1/2 mutation and a non-BRCA1/2 mutation (HOPE_14 had BRCA2 and SPINK1 mutations; HOPE_33 had BRCA2, CDH1, and TP53 mutations; and HOPE_421 had BRCA1 and NBN mutations). Most of the deleterious mutations were found in CDH1 (n=8, 8.4%), RAD51 (n=7, 7.4%), SPINK1 (n=6, 6.3%), TP53 (n=5, 5.3%) and NBN (n=3, 3.2%). The remaining patients had deleterious mutations in CHEK2, FANCA, MLH1 (n=2 of each, 2.1%), BRIP1, MRE11A, MSH2, and MUTYH (n=1 of each, 1.1%).
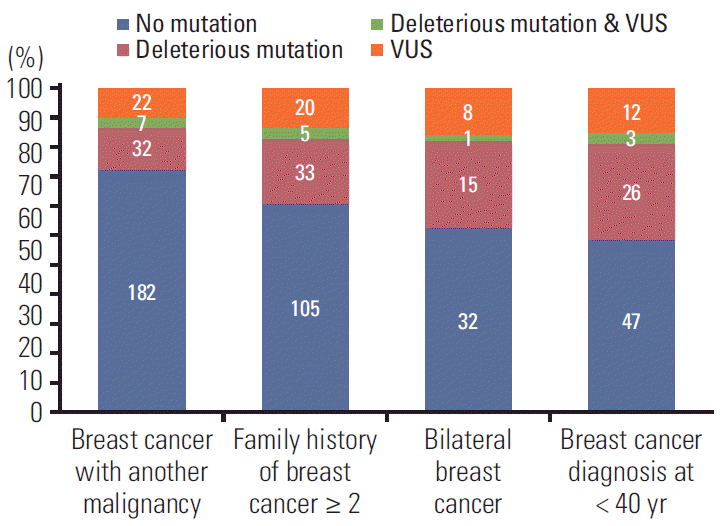
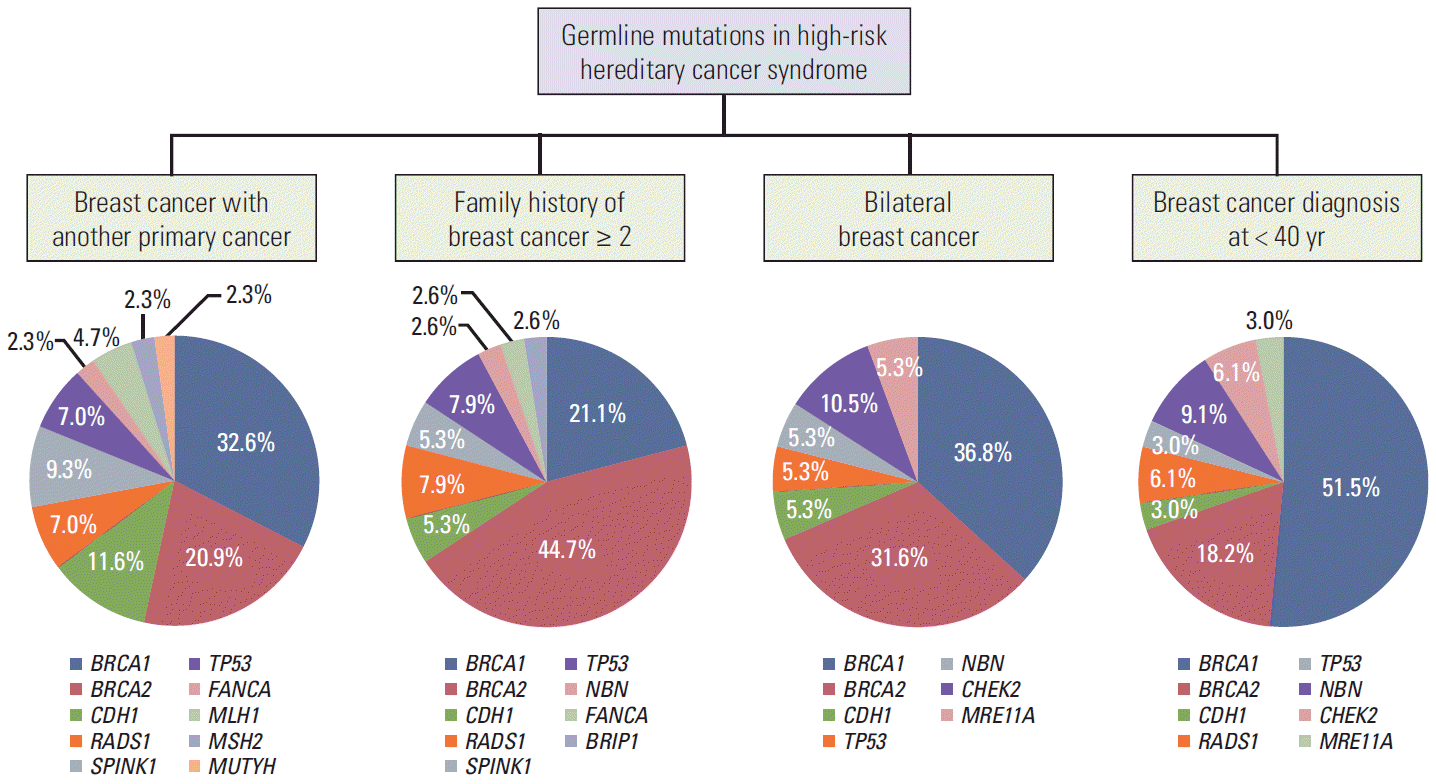
The proportion of deleterious mutations varied according to risk factors. The deleterious mutations were found in 39 of 250 patients (15.6%) who had breast cancer and another primary cancer, 38 of 169 patients (22.5%) who had a family history (≥ 2 relatives) of breast cancer, 16 of 57 patients (28.1%) who had bilateral breast cancer, and 29 of 84 patients (34.5%) who were diagnosed with breast cancer at younger than 40 years old (Fig. 2). Furthermore, the distributions of the cancer susceptibility genes were different according to risk factors (Fig. 3). In breast cancer patients with another primary cancer, BRCA1/2 and non-BRCA1/2 mutations accounted for 52.3% and 47.7% of mutations, respectively. The non-BRCA1/2 mutations comprised CDH1 (11.4%), SPINK1 (9.1%), RAD51 (6.8%), and TP53 (6.8%) mutations. In breast cancer patients with a family history of breast cancer, 65.8% carried a BRCA1/2mutation. In 34.2% of non-BRCA1/2 mutations, 7.9% had RAD51 and TP53 mutations, and 5.3% had CDH1 and SPINK1 mutations. In bilateral breast cancer patients, 68.4% carried a BRCA1/2 mutation. Among the 31.6% who had non-BRCA1/2 mutations, CHEK2 (10.5%) were found frequently and 5.3% of patients had CDH1, TP53, NBN, and MRE11A mutations. In patients diagnosed with breast cancer at younger than 40 years old, 62.1% carried BRCA1/2 mutations and 37.9% carried non-BRCA1/2 mutations including RAD51, NBN, CHEK2, CDH1, TP53, PTEN, FANCA, and MRE11A mutations.
In 64 hereditary cancer-predisposing genes, we found deleterious mutations in 16 genes, including BRCA1/2. However, we did not find deleterious mutations in the remaining 48 genes.
3. Novel deleterious mutations
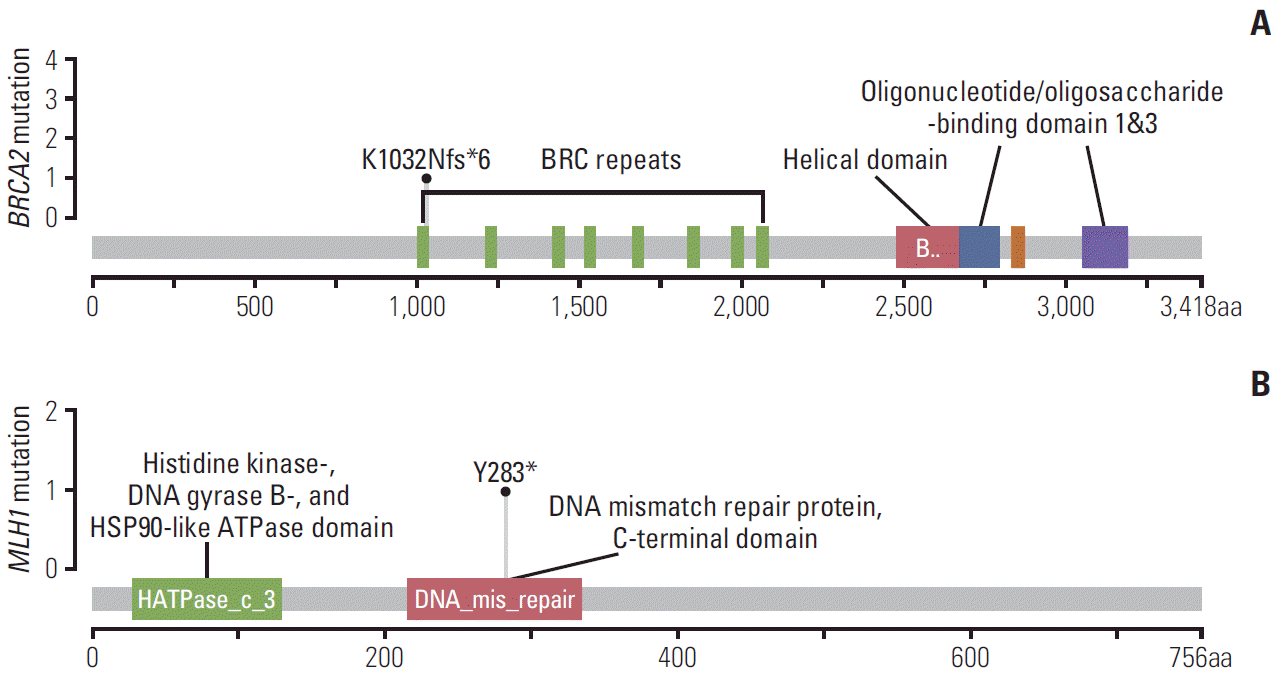
We detected two novel deleterious mutations that were not previously reported: NM_000059.3:c.3096_3111del (p.Lys1032Asnfs*6) in BRCA2 and NM_000249.3:c.849T>A (p.Tyr283*) in MLH1. The NM_000059.3:c.3096_3111del in BRCA2 is identified in patient HOPE_468. This mutation encodes a truncated non-functional protein in the domain of the BRC repeats, interfering with cellular response to DNA damage (Fig. 4A). The NM_000249.3:c.849T>A in MLH1 is identified in patient HOPE_378 and is also predicted to encode a non-functional protein, leading to the disruption of an important functional domain, such as the MutL C-terminal domain (Fig. 4B). The impact of both mutations were predicted deleterious mutations in in silico prediction.
4. Frequency of VUS
A total of 333 missense mutations were identified in 64 genes. After in silico prediction by database and bioinformatics analysis to evaluate pathogenicity, most of the missense mutations were classified as benign or likely-benign. Mutations with conflicting interpretations of pathogenicity but suspicion of being deleterious were classified as VUS. A total of 20 VUS were identified in 67 patients (13.5%) (Table 5). In 15 patients, deleterious mutation and VUS were found concurrently. The proportion of VUS differed among the risk factors for HBOC (Fig. 2). VUS was identified in 11.6% of breast cancer patients with another primary cancer, 14.8% of patients with a family history of breast cancer, 15.8% of bilateral breast cancer patients, and 17.0% of patients who were diagnosed with breast cancer younger than 40 years old. Additionally, 13 patients with VUS also had a concurrent deleterious mutation (HOPE_33, 66, 105, 115, 133, 182, 222, 233, 264, 280, 454, 468, and 501).
Discussion
Patients who carry deleterious mutations are considered to be at high risk for developing cancer, and depending on the target organ, tailored surveillance programs or prophylactic risk-reducing surgery are recommended for decreasing cancer-related mortality. Currently, the National Comprehensive Cancer Network (NCCN) guidelines provide principles of genetic risk assessment and surveillance recommendations for various types of cancer. For example, women with BRCA1/2 mutations are at high risk for breast and ovarian cancers and they are recommended to undergo magnetic resonance imaging (MRI) of the breast for screening and to discuss options for risk-reducing mastectomy or salphingooophorectomy.
We found that 19.2% of breast cancer patients who had clinical features of HBOC had deleterious mutations of cancer susceptibility genes. The breast cancer stage was not different between patients with and without deleterious mutations, nor were the proportions of risk factors for HBOC (breast cancer with another primary cancer, family history of breast cancer in two or more first- or second-degree relatives, and bilateral breast cancer). However, there was a significantly higher rate of patients who were diagnosed with breast cancer at an age younger than 40 years among deleterious mutation-positive patients (p=0.022) (Table 2, Fig. 2). Furthermore, having two or more risk factors for HBOC was also associated with deleterious mutations (p=0.001).
Among 496 patients who were tested by the multiple-gene panel for cancer susceptibility genes, 60 patients (12.1%) were BRCA1/2 positive, which was similar proportion to that reported in Western countries [5]. A previous study that included BRCA1/2-negative Korean breast cancer patients with features of hereditary breast cancer found that only 2.5% of non-BRCA1/2 deleterious mutations were detected: CHEK2 (0.4%), PALB2 (0.9%), MRE11 (0.4%), and RAD50 (0.9%) [6]. Another study including Western patients reported that deleterious mutations were found only in 1.7% of 1994 familial breast cancer patients: PALB2 (1.3%), TP53 (0.3%), CDH1 (0.05%), and ATM (0.05%) [7]. These studies reported that the frequency of deleterious mutations in each gene was less than 1% and concluded that a small portion of hereditary breast cancer was associated with non-BRCA1/2 germline mutations. However, Li et al. [8] detected 11.5% non-BRCA1/2 mutations, including ATM, CDH1, CHEK2, PALB2, PTEN, STK11, and TP53 in 660 cases of familial breast cancer in a Western population. Ricker et al. [3] reported that multiple-gene panel testing increased the detection rate of deleterious mutations from 8.6% to 15.6% compared with a conventional gene-by-gene approach. Furthermore, they repor-ted that there were no significant differences in the mutation rates according to race or ethnic groups [3]. We identified 35 patients (8.0%) who had non-BRCA1/2 deleterious mutations. These mutations included CDH1 in seven patients (1.6%), RAD51 in seven patients (1.6%), SPINK1 in five patients (1.1%), and TP53 in four patients (0.9%). The remaining patients had deleterious mutations in CHEK2 (0.5%), FANCA (0.5%), MLH1 (0.5%), NBN (0.5%), BRIP1 (0.2%), MRE11A (0.2%), MSH2 (0.2%), and MUTYH (0.2%). These results show that multiple-gene panel testing helps to increase the mutation detection rate compared to the conventional BRCA test alone. The results of previous studies and of our study are compatible with the suggestion of NCCN guidelines that multiple-gene testing may be more efficient and cost-effective for cancer risk assessment for patients with a high probability of hereditary cancer syndrome.
Germline CDH1 mutations among the most frequently detected deleterious non-BRCA1/2 mutations in our study. CDH1 mutation is known to be associated with invasive lobular carcinoma and diffuse gastric cancer [9,10]. The NCCN guidelines recommend that women with the CDH1 mutation receive regular breast examinations with annual mammogram and breast MRI, as well as prophylactic total gastrectomy or regular esophagogastroduodenoscopy with multiple random biopsy. In this study, we found eight patients with CDH1 mutation (NM_004360.4:c.2494G>A), 7 patients with CDH1 mutation only, and one patient (HOPE_33) with CDH1 and another mutations (BRCA2 NM_000059.3:c.3195_3198del and TP53 NM_000546.5:c.566C>T). Patient HOPE_33 carried BRCA2, CDH1, and TP53 mutations; she developed bilateral breast cancer at 33 years of age. Two patients with only the CDH1 mutation had a family history of breast cancer in two or more first- or second-degree relatives. Five patients with only the CDH1 mutation had breast cancer with another primary cancer including leukemia (HOPE_23), stomach cancer (HOPE_28), colon cancer (HOPE_222), cervical cancer (HOPE_192), and thyroid cancer (HOPE_319). Patients with the CDH1 mutation should have been recommended to receive close surveillance for contralateral breast cancer and stomach cancer. Further, family members of patient HOPE_28, who already had stomach cancer, should undergo genetic testing and receive close surveillance for breast and stomach cancers.
The RAD51 gene has a key role in the repair of DNA double-strand breaks through homologous recombination [11]. Germline mutation of RAD51 is known to cause congenital mirror movement which is characterized by involuntary movements of one side of the body that mirror intentional movements on the opposite side [12]. In addition to this congenital neurologic disorder, RAD51 mutation is associated with the development of malignancy, in including breast cancer and pancreatic cancer [13,14]. A previous study found that the proteins BRCA2 and PALB2 control the function of RAD51, yielding structural change for cancer susceptibility [15]. In this study, we found eight patients with RAD51 NM_002875.4:c.449G>A. All patients with RAD51 mutation were BRCA1/2 negative. Three patients (HOPE_24, 35, and 335) had breast cancer and another primary cancer, including lung cancer, brain tumor, and thyroid cancer. Another three patients (HOPE_231, 324, and 418) had a family history of breast cancer in two or more relatives. One patient (HOPE_266) had breast cancer at an age younger than 40 years.
Germline mutation of SPINK1 has been associated with hereditary pancreatitis by inhibiting the function of SPINK1 protein and causing cellular damage by activated trypsin [16]. Several studies found that the SPINK1 mutation was associated with pancreatic cancer [17]. In our study, 6 patients carried the deleterious SPINK1 mutations (NM_003122.4:c.101A>G). One patient (HOPE_14) had both breast cancer and pancreatic cancer and found to carry both SPINK1 and BRCA2 mutations (NM_000059.3:c.4092_4093del). Another three patients had breast cancer and an additional primary cancer, including stomach cancer, cervical cancer, and common bile duct cancer. The remaining two patients had a family history of breast cancer. However, SPINK1 mutation is not rare despite of deleterious mutation. According to 1000 Genome Project Phase 3, allele frequency of this mutation is 0.003 in American, 0.008 in East Asian and 0.014 in South Asian. Because the allele frequency is relatively high in Asian population, this mutation is thought to have low penetrance in Asian population. Patient HOPE_14 who carried SPINK1 and BRCA2 mutations developed breast cancer in 2002 and pancreatic cancer in 2005. Considering that the BRCA2mutation is also known for increasing risk of pancreatic cancer and high minor allele frequency of SPINK1 mutation (NM_003122.4:c.101A>G), the main cause of breast and pancreatic cancer in patients HOPE_14 was BRCA2 mutation, not SPINK1 mutation [18].
Germline mutation of TP53 is known as Li-Fraumeni syndrome. This mutation is associated with multiple cancers including breast cancer, soft tissue sarcoma, acute leukemia, brain tumor, adrenal carcinoma, and colon cancer. For this reason, Li-Fraumeni syndrome patients have a poor prognosis. Once this syndrome was revealed to be associated with a germline mutation of TP53, it became possible to detect carriers of inherited TP53 mutations. Currently, individuals with a TP53 mutation are recommended to undergo targeted surveillance, depending on individual medical history and family history. Villani et al. [19] reported that individuals with TP53 mutation who received intensive surveillance with colonoscopy, whole body MRI, breast MRI, brain MRI, skin examination, and physical examination showed improved overall survival compared with individuals who did not receive surveillance (p=0.013). This result supports the effectiveness of a tailored surveillance program for increasing survival rates and is beneficial to individuals with deleterious mutations. In our study, five patients were identified to carry TP53 mutations (NM_000546.5:c.566C>T, NM_000546.5:c. 638G >A, and NM_000546.5:c.743G>A). As mentioned, HOPE_33 carried BRCA2, CDH1, and TP53 mutations and had bilateral breast cancer at a young age. HOPE_395 and 396 carried TP53 mutation (NM_000546.5:c.566C>T) and suffered from breast cancer and thyroid cancer and had at least two relatives with a history of breast cancer. HOPE_290 had TP53 mutation (NM_000546.5:c.638G>A) and a family history of breast cancer in at least two relatives. HOPE_115 had TP53 mutation (NM_000546.5:c.743G>A) and had breast cancer and lung cancer. Family members of patients with TP53 mutation need to undergo genetic testing to find out whether they are carriers of the TP53 mutation or not. Depending on the results of genetic testing, TP53 mutation carriers, as well as patients with TP53 mutation, should consider clinical intensive surveillance for early detection of cancer and improved long-term survival.
Surveillance and risk-reducing strategies for patients with germline mutations of Lynch syndrome (MLH1, MSH2, MSH6, PMS2, and EPCAM2), such as colonoscopy, prophylactic hysterectomy, and bilateral salphingo-oophorectomy should be considered. In our study, three patients were found to have deleterious mutations in MLH1 and MSH2 (NM_000249.3: c.1758dup, NC_000003.11:c.849T>A, and NM_000251. 2:c.229_230del). Patient HOPE_315 who carried MLH1 mutation (NM_000249.3:c.1758dup) had primary breast, colon cancer, and lung cancer. Patient HOPE_394 who had MSH2 mutations (NM_000251.2:c.229_230del) had primary breast and colon cancers. Patient HOPE_378 who carried novel deleterious mutation in MLH1 (NM_000249.3:c.849T>A) had breast cancer and hepatocellular carcinoma. Because Lynch syndrome is an inherited disorders that increases the risk of various type of cancer, particularly in colorectum, endometrium, ovary, stomach, small bowel, liver, bile duct, upper urinary tract, and brain, these three patients’ family members should have intensive surveillance for colon cancer and genetic testing for germline mutations of Lynch syndrome.
It is well known that BRCA1 and BRCA2 mutation carriers showed differences in tumor histopathology. A large proportion of breast cancer in women who carry a BRCA1 mutation exhibited a triple-negative breast cancer. Previous study including Korean familial breast cancer patients reported that triple-negative breast cancer was diagnosed in 57.1% of BRCA1 mutation carriers [20]. In contrast with BRCA1 mutations, luminal A and luminal B subtype of breast cancer was found in 83.0% in breast cancer patients who carried BRCA2 mutations [21]. Our results showed that 75.0% of BRCA1 mutation carriers were triple-negative breast cancer and 69.5% of BRCA2 mutation carriers were HR-positive breast cancer including luminal A and B subtype, which are concordant with previous studies (Table 3).
The proportions and distributions of deleterious mutations in BRCA1/2 negative patients were quite different in this Asian population than in a previously reported Western population. Maxwell et al. [22] reported that Caucasian and African American breast cancer patients who were BRCA1/2 negative and had early-onset breast cancer (< 40 years old at diagnosis) carried 11% of non-BRCA1/2 deleterious mutations. The deleterious mutations were ATM (25.8%), CHEK2 (32.3%), TP53 (12.9%), and MRE11A (6.5%). The remaining mutations were MSH6, CDKN2A, MUTYH, BARD1, BRIP1, NBN, and RAD50 (3.2%). The majority of deleterious mutations in our study in BRCA1/2-negative and early-onset breast cancer patients were NBN (30.0%), RAD51 (20.0%), and CHEK2 (20.0%) (Fig. 3). Recently, Li et al. [23] reported the results of germline mutations among Chinese patients with features of hereditary breast cancer. They found that 16.9% of included patients carried BRCA1/2 mutations and 6.8% of patients had non-BRCA1/2 mutations including TP53, PALB2, CHEK2, ATM, BARD1, BRIP, CDH1 and RAD50. Recent studies reported that mutations in PALB2 and RAD51C were found to be an important cause of HBOC [24,25]. Additionally, CDH1 mutations were not found in the Western study but detected in the Chinese study. Although we did not find the ATM, PALB2, and RAD51C mutations in any of our study patients, we should have caution to interpret sequencing results of these important genes for HBOC.
We detected 2 novel deleterious mutations that have not been previously reported: NM_000059.3:c.3096_3111del (p.Lys1032Asnfs*6) in BRCA2 and NM_000249.3:c.849T>A (p.Tyr283*) in MLH1. The p.Lys1032Asnfs*6 mutation in BRCA2 was identified in patient HOPE_468. This mutation encodes a truncated non-functional protein in the domain of the BRC repeats (Fig. 4A). The human tumor suppressor protein BRCA2 plays a key role in recombinant DNA repair. BRCA2 recruits RAD51 to sites of DNA damage through interaction with eight conserved motifs of approximately 35 amino acids, the BRC repeats, although the specific function of each repeat remains unclear [26]. The mutation of BRCA2 p.Lys1032Asnfs*6 is thought to interfere with cellular response to DNA damage, resulting in malignant transformations. The p.Tyr283* mutation in MLH1 is found in patient HOPE_378 and is also predicted to encode a non-functional protein, leading to the disruption of important functional domain like MutL C-terminal domain (Fig. 4B). The subunits of MLH1 and PMS2 make the MutLα complex, which plays an essential role in mismatch repair [27]. A defect in MLH1 is associated with mismatch repair and results in microsatellite instability and spontaneous mutation rate. The family history of patients with novel deleterious mutations and genetic tests of family members are required to determine the clinical impact of these newly identified mutations.
In this study, 67 patients (13.5%) were shown to have 20 VUS in 18 genes (Table 5). Compared with other studies, the rate of VUS in this study was relatively low. This could be because we excluded most of the missense mutations with conflicting interpretations and considered benign or likely-benign. We only considered mutations as VUS when mutations had conflicting interpretation of pathogenicity but a suspicion of being deleterious. Most of the VUS will be re-categorized as benign or deleterious. Until the significance is fully understood, VUS should not be used for making clinical decisions. It is also important to reduce the number of VUS in clinical practice. Potential deleterious mutations can be selected by mutation frequency analysis and in silico analysis. Recently, Findlay et al. [28] used saturation genome editing to assay single-nucleotide variants in exons that encode functional domains of BRCA1. They found that functional effects of saturation genome editing were almost perfectly concordant with established assessments of pathogenicity. The saturation genome editing will be useful for accurate classification of VUS in clinically actionable genes.
The application of multi-gene panel testing has been rapidly increasing in clinical practice, especially in the evaluation of germline mutations which are associated with cancer susceptibility. The identification of deleterious mutations in cancer susceptibility genes in individuals with a high risk for hereditary cancer can improve the effectiveness of personalized surveillance, leading to early detection or prophylactic treatment of hereditary cancer in both individuals and their family members. Intensive surveillance for early detection and prophylactic treatment is directly linked with better survival in patients with deleterious mutations.
However, there are limitations to multi-gene panel testing. The prevalence of pathogenic mutations and VUS vary across races and ethnicities. Furthermore, the penetrance and phenotype of mutations are different among individuals. Detection of a deleterious mutation does not always mean an individual will develop cancer, and conversely, a negative result from a multi-gene panel test does not mean an individual has no risk of getting cancer. Although two novel mutations were found to be pathogenic because of their functioning effect on protein level in in silico analysis, theses mutations were not clinically verified. Additional investigation including family history and targeted genetic tests of family members are required to determine the clinical impact of these newly identified mutations.
Another limitation is that we could not compare sequencing results between NGS and Sanger sequencing with same blood sample because of shortage of sample amount. The NGS-based multi-gene panel testing have weakness point in detecting mutations compared with Sanger sequencing including uncovered area, large insertion/deletion, and copy-number variation. Although these weaknesses can be overcame by technical improvement, clinical implication including long-term outcomes should be discussed carefully.
Last limitation is that the attitude and knowledge gaps of physicians who provide care for individuals who undergo genetic testing for a disease. One survey reported that, although most physicians received formal genetic education and agreed that genetic tests are clinically useful for assessing disease risk, they were not confident about interpreting test results and were not prepared for managing individuals at high risk for genetic disease. For these reasons, genetic education and genetic counseling, as well as the appropriate and accurate interpretation of results, are important for the effective clinical application of risk management strategies. Stadler et al. [29] proposed that the results of germline genetic testing using multi-gene panels, including cancer-related findings and other incidental findings, should be integrated with traditional risk assessments, such as personal and family histories, to establish cancer and non-cancer risk management and follow-up plans. The paradigm shift toward personalized and precision medicine requires the incorporation of NGS technologies into clinical practice.
To the best of our knowledge, this is the largest study to include Korean breast cancer patients with clinical features of HBOC and examine the frequency and characteristics of germline mutations in BRCA1/2 and non-BRCA1/2 cancer susceptibility genes.
We analyzed germline mutations from 496 breast cancer patients of Asian ethnicity with clinical features of HBOC using NGS-based multi-gene panel testing. Overall, 95 patients (19.2%) were found to carry 48 deleterious germline mutations in 16 cancer susceptibility genes. Of these 95 patients, 60 patients (63.2%) had BRCA1/2mutations, 38 patients (40.0%) had non-BRCA1/2 mutations and three patients (3.2%) had both BRCA1/2 and non-BRCA1/2 mutations. The NGS-based multi-gene panel test improved the detection rates of deleterious mutations and provided a cost-effective cancer risk assessment compared with a gene-by-gene approach.
 XML Download
XML Download