

 PDF
PDF Citation
Citation Print
Print


Introduction
Epstein-Barr virus (EBV) was the first virus shown to encode viral microRNAs (miRNAs, miR). Five EBV-associated miRNAs were firstly identified from EBV B95-8–infected Burkitt’s lymphoma in 2004 by Pfeffer et al. [1]. To date, 44 mature miRNAs that could be classified into two clusters (BHRF1 cluster and BART cluster) have been identified in EBV. BHRF1 miRNA is highly expressed in stage III latency while BART miRNAs are expressed in all EBV-positive cell lines [2]. Their contributions to viral pathogenesis and oncogenesis have been shown in many EBV-associated lymphomas [3,4]. Diverse cellular pathways including p53, B-cell signaling, immune activation, cell proliferation, and apoptosis are now known to be regulated by EBV miRNAs in lymphomas [5].
Our previous study showed that nearly 10.0% of newlydiagnosed chronic lymphocytic leukemia (CLL) patients had a high level of EBV-DNA viral load in whole blood [6]. Furthermore, a positive blood EBV-DNA test at diagnosis was significantly associated with inferior time-to-treatment (TTT) and overall survival (OS) in CLL patients [6,7]. Further studies are needed to investigate the potential mechanisms of EBV’s oncogenesis in CLL patients.
p53 gene has a vital role in the disease-progress, clone evolution and drug-resistance for CLL patients. Until now, no therapies can fully overcome the inferior prognosis caused by p53 aberration even in the era of the emerging of so many molecular targeted drugs such as BTK inhibitor and Bcl-2 inhibitors [8-10]. Interestingly, one of the members of the BHRF1 miRNAs, EBV-miR-BHRF1-1, was found to bind to the 3′-untranslated regions (UTR) of p53, suggesting that p53 may be a target of EBV-miR-BHRF1-1 [11]. In our studies, we provide functional evidence that p53 is indeed a cellular target for EBV-miR-BHRF1-1. We demonstrated that the inhibition of p53 expression by EBV-miR-BHRF1-1 is likely to confer a growth advantage to EBV-infected CLL cells. These findings provide one of the potential mechanisms by which EBV can promote CLL development through its miRNA functions.
Materials and Methods
1. Subjects
We collected 97 previously-untreated CLL patients from January 2004 to December 2015. The diagnosis of CLL was based on the revised National Cancer Institution criteria [12]. Variables ascertained at diagnosis included:age, sex, Binet stage, blood absolute lymphocyte level (ALC), lactate dehydrogenase (LDH), β2-microglobulin, EBV-DNA load in whole blood, IGHV mutation state, p53 aberration (p53 mutation or deletion) state and cytogenetics (studied by fluorescence in situ hybridization).
2. Lentivirus transfection
The cell lines we used for cell function experiments in vitro were MEC1 and JVM3. Cells were plated in 6-well plates at a density of 2.0×105 cells/well and stably transfected with maturing miRNA inhibitors of EBV-miR-BHRF1-1, maturing miRNA mimics of EBV-miR-BHRF1-1, and negative control random miRNA, respectively (GeneChem, Shanghai, China), using enhanced infection solution and polybrene (GeneChem) according to the manufacturer’s protocol. The lentivirus was loaded with gene that contains green fluorescence and puromycin resistance gene. The 6-well plates were centrifuged by centrifugal machine (Eppendorf Centrifuge 5810 R, Hamburg, Germany), 2,000 rpm, 30 minutes at 37°C. The cell medium (IMDM) was changed after 3 days of lentivirus transfected and puromycin (1 μg/mL) was added into a new medium to screen the transfected cells. The cell medium (IMDM) was changed after another 3 days and cultured cells for further function experiments.
3. EBV-miR-BHRF1-1 detection
Total RNA was extracted from CLL patients’ peripheral blood mononuclear cells, cultured cell lines and lentivirus transfected cell lines. RNA (1 μg) was reverse transcribed to cDNA by using specific stem-loop reverse transcription primers, forward primers, reverse primers (S1 Table), and amplification with fluorescent dye SYBR Green MasterMix by using reverse transcription kit (GenePharma, Shanghai, China). U6 was used as an internal control. Cycle conditions for EBV-miR-BHRF1-1 and U6 were one cycle for 3 minutes at 95°C, 40 cycles for 12 seconds at 95°C, 40 seconds at 62°C, and melting curves were measured at the same time. The threshold cycle (Ct) was defined as the cycle number at the fluorescence over the fixed threshold. ΔCt was calculated by subtracting the Ct of U6 from the Ct of EBV-miR-BHRF1-1. Relative expression level of a gene was calculated by the equation 2–ΔCt. Reactions for quantitative reverse transcription–polymerase chain reac-tion (qRT-PCR) were conducted in triplicate, using the App-lied Biosystems ABI StepOnePlus Real-time PCR system (Applied Biosystems, Foster City, CA) and replicated for three times. Productions were verified by DNA sequencing.
4. p53 mRNA detection
Total RNA was extracted from cultured cell lines and lentivirus transfected cell lines. RNA (1 μg) was reverse transcribed using random hexamers, and amplification with fluorescent dye SYBR Green MasterMix and primers (S1 Table). β-Actin was used as an internal control. Cycle conditions for p53 and β-actin were one cycle for 5 minutes at 95°C, 35 cycles for 15 seconds at 95°C, 30 seconds at 60°C, 30 seconds at 72°C and melting curves were measured after amplification. ΔCt was calculated by subtracting the Ct of β-actin from the Ct of p53. ΔΔCt was calculated by the ΔCt of unprocessed cell lines from the ΔCt of lentivirus transfected cell lines. Relative expression level of a gene was calculated by equation 2–ΔΔCt. Reactions for qRT-PCR were conducted three times, using the Applied Biosystems ABI StepOnePlus Real-time PCR system (Applied Biosystems). Productions were confirmed by DNA sequencing.
5. Dual-luciferase reporter assay
Renilla-luciferase assay was performed to verify whether EBV-miR-BHRF1-1 is combined with p53 gene. The pmirGLO Dual-Luciferase miRNA Target Expression Vector is designed to quantitatively evaluate miRNA activity by the insertion of miRNA target sites 3´-UTR region of the firefly luciferase gene (luc2). EBV-miR-BRHF1-1 or negative controls (NC) were transfected together with pmirGLO vector in the 293T cell line by using lipofectamine 2000 (Invitrogen, Carlsbad, CA). The ratios of Renilla versus firefly signals was used to measure the efficiency of transfection by using dualluciferase reporter gene assay kit (Promega, Madison, WI) at 24 hours and 48 hours after transfection.
6. Western blotting
Cells were harvested after stable transfection with miRNA inhibitors (including EBV-miR-BHRF1-1 and NC miRNA inhibitors) and miRNA mimics (including EBV-miR-BHRF1-1 and NC miRNA mimics). CLL patients’ peripheral blood mononuclear cells and cells of transfection were and solubilized by incubating in ice-cold RIPA lysis buffer. Then cells were fragmented fully by ultrasonic instruments. Protein concentration was determined by BCA (Real-Times, Shanghai, China). Proteins were added in sodium dodecyl sulfatepolyacrylamide gel (SDS-PAGE) loading buffer (Beyotime, Shanghai, China) for dye and storage. Protein samples separated on 12% SDS-PAGE (Beyotime), transferred to polyvinylidene difluoride membranes (Immobilon-P Membrane) followed by incubating with a mouse monoclonal antibody to p53 (Cell Signaling Technology, Boston, MA) at 1:1,000 dilutions, a rabbit monoclonal antibody to p-p53 (Cell Signaling Technology) at 1:1,000 dilutions, a rabbit monoclonal antibody to PDLIM7 (Cell Signaling Technology) at 1:1,000 dilutions, and a rabbit monoclonal antibody to p21 (Cell Signaling Technology) at 1:1,000 dilutions. The secondary antibody used was rabbit anti-goat IgG (Multi Sciences) and mouse anti-goat IgG (Multi Sciences) both at 1:3,000 dilutions at room temperature. Protein levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology), a mouse monoclonal antibody, at 1:10,000 dilutions. Antibody complexes were detected by enhanced chemiluminescence (Merck Millipore, Darmstadt, Germany).
7. Cell Counting Kit-8 proliferation experiment
Cells were plated in 96-well plates at a density of 1.0×104 cells/well with 100 μL IMDM medium, and stabilized cells for 4 hours at 5% CO2, 37°C incubator (Thermo Fisher Scientific, Boston, MA). Then take out to add in 10 μL Cell Counting Kit-8 (CCK-8) at once, 24, 48, 72, and 96 hours after planked cells in triplicate, using CCK-8 kit (Dojindo, Kumamoto, Japan) and incubate cells continuously. After 2 hours, OD value was tested by using a microplate reader (Bio-Tek, Winooski, VT) and data were collected by Gene5 software (Bio-Tek).
8. Apoptosis assay
Cells after stable transfection was performed to detect apoptosis ratio by using Annexin V propidium iodide (PI)/ 7-aminoactinomycin D (7-AAD) flow cytometric assay kit (BD Pharmingen, Franklin Lakes, NJ). We collected 1.0×106 transfected cells into a tube, and cells were washed twice with cold phosphate-buffered saline (PBS) and re-suspended in 1 mL Annexin V binding buffer. Cells were incubated in the dark for 15 minutes at room temperature after the addition of 5.0 μL of PI and 3.0 μL of 7-AAD. We added 4 mL binding Annexin V buffer additionally before test. Apoptosis of lentivirus transfected cells was quantified using a FACSCalibur flow cyto-meter (BD FACSCanto II Flow Cytometer) and FlowJo software. Phycoerythrin and PI are the same fluorescent emission channel. 7-AAD and PerCP-Cyanine5- 5 (PerCP-cy5-5) are the same fluorescent emission channel. Experiments were done in triplicate.
9. Cell cycle analysis
Cells at the concentration of 1.0×106 were incubated and saved at –20°C for 30 minutes fixed in 70% ethanol, then washed twice in PBS and incubated in 1 mL DNA staining solution and 10 μL PI for 30 minutes at 37°C using cell cycle staining kit (MultiSciences). DNA content was determined by flow cytometry using Beckman Coulter Gallios flow cytometry (Boston, MA) and analyzed by Wincycle 32 software (Beckman Coulter). Experiments were done in triplicate.
10. Statistical analyses
Statistical analyses were performed using SPSS software for Windows ver. 20.0 (IBM Corp., Armonk, NY). The difference of target gene mRNA expression between groups with different prognostic factors was described using the MannWhitney U test. The difference of miRNA and p53 expression and apoptosis rate between groups was described using the paired-samples T test. The receiver operating characteristic (ROC) curve was used to determine the cut-off value. OS was calculated as time from diagnosis until death or last followup. TTT was calculated as interval from diagnosis until the first CLL-specific treatment or the last follow-up. Survival and TTT were estimated by the Kaplan-Meier method and results were compared using the log-rank test. The prognostic influence of variables was tested using the Cox proportional hazards model in univariate and multivariate analysis. Protein bands of Western blot were quantified through the Image J program for Windows after normalizing the data for GAPDH. Two-sided p-values of < 0.05 were considered statistically significant.
Results
1. Clinical variables
The baseline clinical and biological variables of the 97 newly-diagnosed CLL patients are shown in Table 1. The median age at diagnosis was 59 years (range, 16 to 82 years). There were 64 males (65.9%). Sixty-one patients (62.8%) were Binet stage A and 36 (37.1%) patients were staged as B/C. There were 21 patients (21.6%) who had pretreatment high EBV-DNA copies (more than 5,000 copies/mL) in whole blood. Thirty-two (33.0%) had lymphocyte count of more than 50×109/L and 31 patients (32.0%) had elevated LDH levels. Thirty-five patients (36.1%) were IGHV gene unmutated. p53 aberration (p53 mutation or deletion) was detected in 22 of 97 subjects (22.7%) and 10 patients (10.3%) had both p53 mutation and deletion. The median duration of follow-up is 71 months (range, 36 to 138 months). Seventy patients (72.2%) had indications and received treatment. Seventeen (17.5%) subjects died of CLL-related causes.
2. EBV-miR-BHRF1-1 expression and association with other variables
The relative expression level of EBV-miR-BHRF1-1 in all subjects was 0.00001 to 0.05120 (median, 0.01056). EBV-miRBHRF1-1 expression of CLL subjects was analyzed for association with baseline variables (Table 1). Significant associations were found with LDH ≤ upper limit of normal (ULN) vs. LDH > ULN (p=0.007), IGHV mutation vs. IGHV unmutation (p < 0.001) and p53 disruption vs. no disruption (p < 0.001).
3. The prognostic roles of EBV-miR-BHRF1-1 in CLL patients
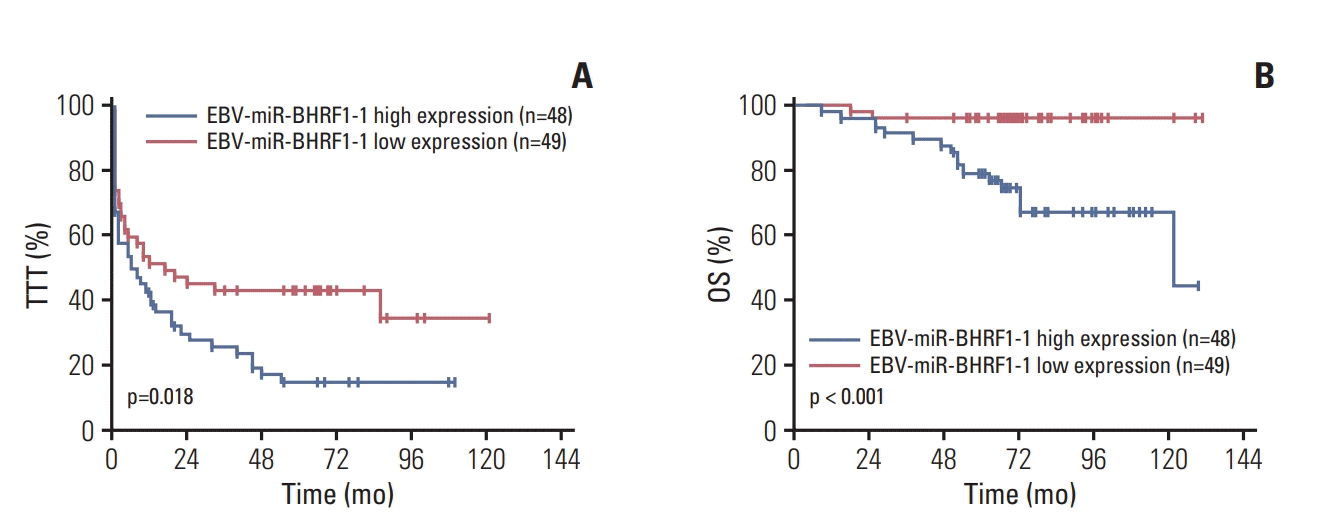
ROC curve analysis of OS was used to determine the optimal threshold cut-off value for EBV-miR-BHRF1-1 level. Area under the curve was 0.651 (95% confidence interval [CI], 0.531 to 0.771; p < 0.001). The optimal cut-off value of EBV-miR-BHRF1-1 was 0.0012, with a 51% sensitivity and a 77% specificity. We split the cohort of CLL into low (n=49) and high (n=48) EBV-miR-BHRF1-1 expression based on a value of 0.0012. Subjects in the low EBV-miR-BHRF1-1 expression cohort had a briefer TTT (p=0.018) and worse OS (p < 0.001) compared with subjects in high EBV-miR-BHRF1-1 expression cohort (Fig. 1).
In univariate analyses, Binet B/C stage, ALC ≥ 50×109/L, LDH > ULN, EBV-DNA > 5,000 copies/mL, IGHV unmutated, p53 disruption, EBV-miR-BHRF1-1 high expression were significantly associated with inferior TTT (Table 2). However, in multivariate analyses, only ALC ≥ 50×109/L (p=0.030; hazard ratio [HR], 1.960; 95% CI, 1.068 to 3.599), LDH > ULN (p=0.046; HR, 1.695; 95% CI, 1.009 to 2.848) and IGHV unmutated status (p=0.024; HR,1.828; 95% CI, 1.083 to 3.087) remained significant (Table 2).
In univariate analyses, LDH > ULN, IGHV unmutated, p53 disruption and EBV-miR-BHRF1-1 high expression were significantly associated with inferior OS (Table 2). However, in multivariate analyses, only IGHV unmutated (p=0.023; HR, 4.433; 95% CI, 1.223 to 16.069), p53 disruption (p=0.013; HR, 2.316; 95% CI, 1.197 to 4.482), and EBV-miR-BHRF1-1 high expression (p=0.028; HR, 5.335; 95% CI, 1.193 to 23.846) remained significant (Table 2).
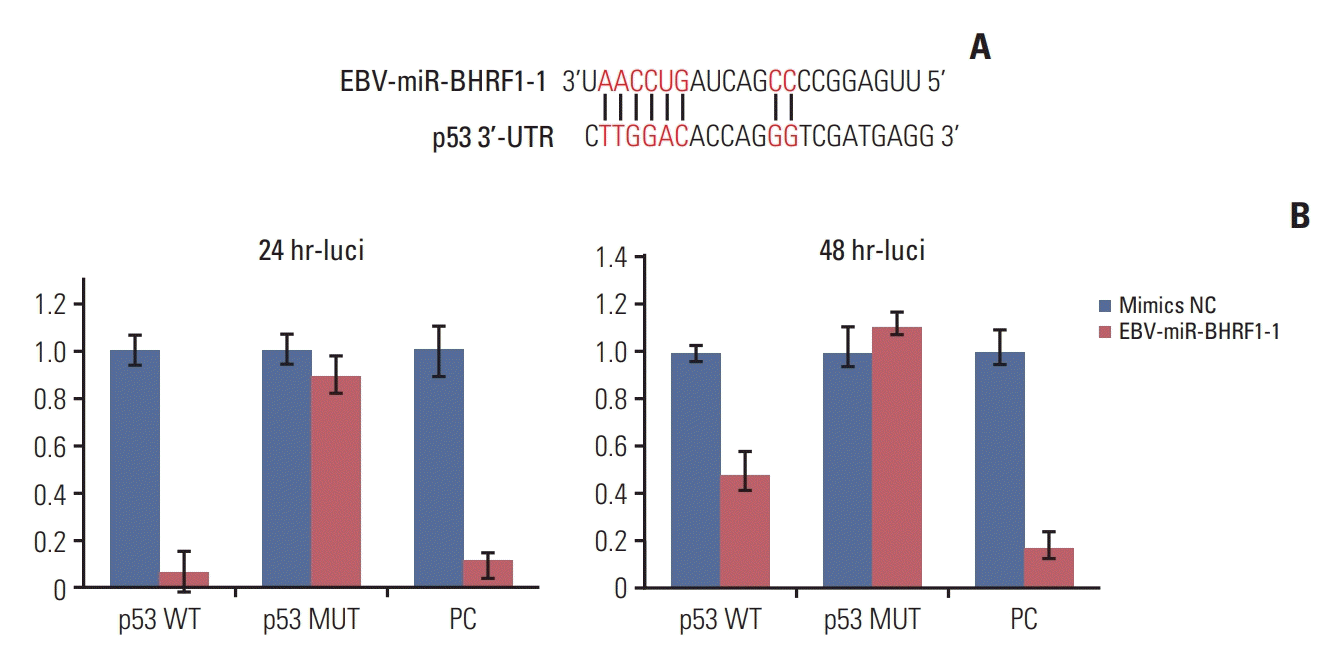
4. EBV-miR-BHRF1-1 targets the 3’UTR of p53 mRNA
The computational algorithms, including TargetScan and RNAhydrid, were used in combination to identify potential miRNAs that target p53. As shown in Fig. 2A, the bioinformatics analysis identified EBV-miR-BHRF1-1 as the possible regulator of p53. As a first step to validate that p53 is indeed a functional target of EBV-miR-BHRF1-1 mediated through the potential miRNA blinding site, we used luciferase reporters containing either the wild-type p53 3′-UTR harboring a point mutation in the critical seed region of the EBV-miR-BHRF1-1 binding site. Wild-type or mutant reporter plasmid was then co-transfected with 100 nM EBV-miR-BHRF1-1 mimics into 293T cells, and their luciferase activities were measured at 24 hours and 48 hours. EBV-miR-BHRF1-1 repressed wild-type p53 luciferase reporter activity by 6.28% at 24 hours (p < 0.001) and 48.24% at 48 hours (p < 0.001) but had no effect on the luciferase activity of the mutant p53 plasmid (Fig. 2B). Thus, the current results showed that EBV-miR-BHRF1-1 inhibits the expression of p53 by binding to the target sequence.
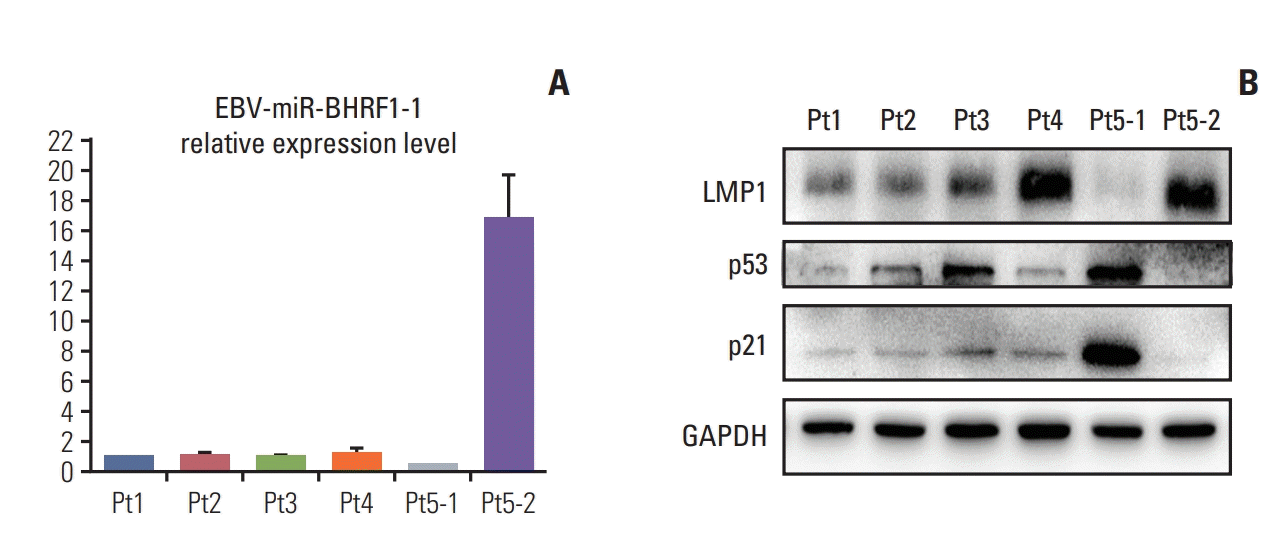
5. High expression of EBV-miR-BHRF1-1 correlates with low p53 expression in EBV-positive CLL
To further investigate whether there is also a negative correlation between EBV-miR-BHRF1-1 levels and p53 expression in primary EBV-positive CLL cases, EBV-miR-BHRF1-1 and p53 expression were assessed in five CLL cases (Fig. 3). Patient 4 has a relatively higher level of EBV-miR-BHRF1-1 and latent membrane protein 1 (LMP1) protein expression and lower level of p53 and p21 protein expression. Patients 1, 2, and 3 showed relatively lower levels of EBV-miR-BHRF1-1 and LMP1 protein expression and higher levels of p53, and p21 protein expression. This negative association between EBV-miR-BHRF1-1 and p53 expression is well demonstrated in patient 5, whose samples patient5-1 (Pt5-1) and patient5-2 (Pt5-2) represent the initial diagnostic and relapsed samples, respectively. When the disease of case 5 relapsed after standard treatment, the levels of EBV-miR-BHRF1-1 increased to relatively higher level and LMP1 protein. Furthermore, we found that the levels of p53 and p21 decreased to relatively lower level.
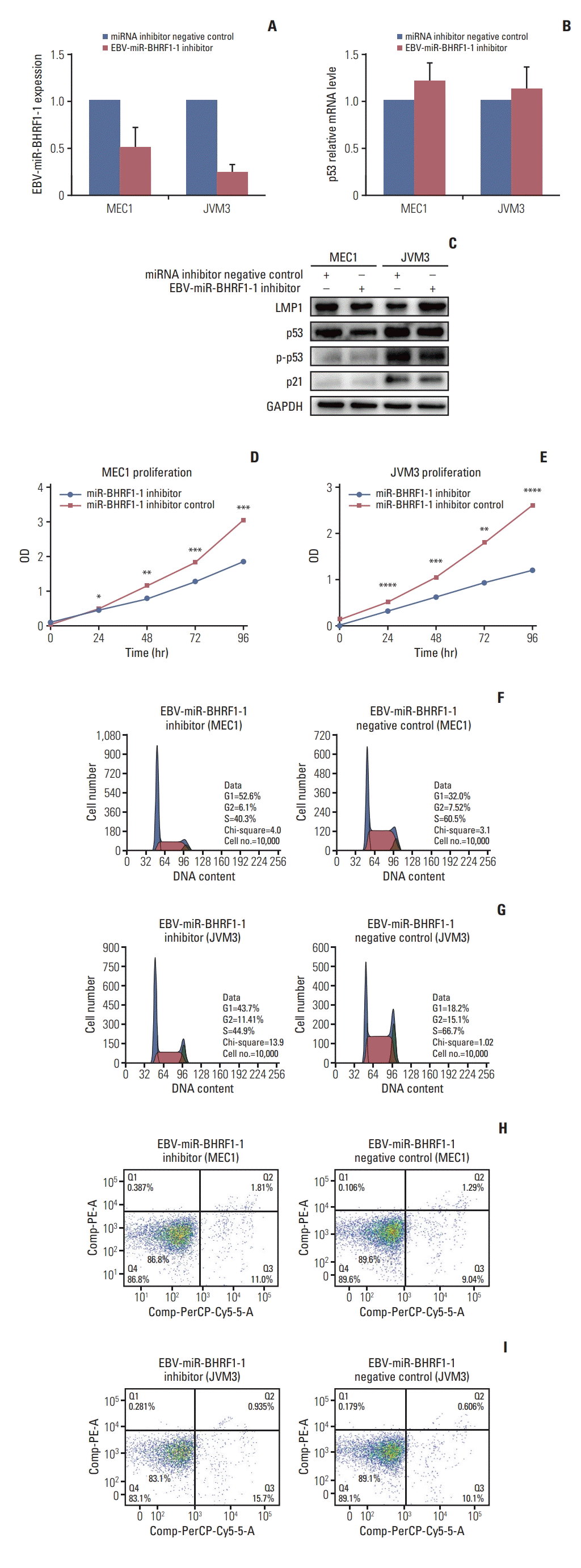
6. EBV-miR-BHRF1-1 inhibitor up-regulate p53 expression and affected CLL cell functions in MEC1 and JVM3
We then explored the biological effects of inhibiting EBV-miR-BHRF1-1 on cell lines of MEC1 and JVM3 and whether of these effects may be mediated through increase of p53 levels. We only used EBV-miR-BHRF1-1 inhibitor to decrease the EBV-miR-BHRF1-1 levels in cell lines. qRT-PCR, western blotting (WB) and cell function tests were conducted after cells were transfected stable. Significant reductions of EBV-miR-BHRF1-1 expression level were observed in both MEC1 and JVM3 (Fig. 4A). Although p53 mRNA level remained unchanged on EBV-miR-BHRF1-1 inhibitor transfection compared with NC (Fig. 4B), WB revealed up-expression level of p53, p-p53 and p53 proteins (Fig. 4C). The CCK-8 experiments were done to detect the proliferation ability of cell in 24, 48, 72, and 96 hours. Lower proliferation ability was observed in MEC1 and JVM3 cell lines transfected with EBV-miR-BHRF1-1 inhibitor compared with MEC1 and JVM3 cell lines transfected with miRNA NC (Fig. 4D and E). This is associated with a decrease in cell cycle progression based on BrdU incorporation assay. As shown in Fig. 4F and G, the S phase was reduced from 60.5% in MEC1 cells and 66.7% in JVM3 cells transfected with miRNA inhibitor NC, to 40.3% and 44.9% on EBV-miR-BHRF1-1 inhibitor transfection (p < 0.05). Apoptosis assay was performed to ascertain the biological effect of EBV-miR-BHRF1-1 inhibitor on the apoptosis of cell lines. A marked increase in apoptosis was observed in the MEC1 and JVM3 cell lines transfected with EBV-miRBHRF1-1 inhibitor (Fig. 4H and I).
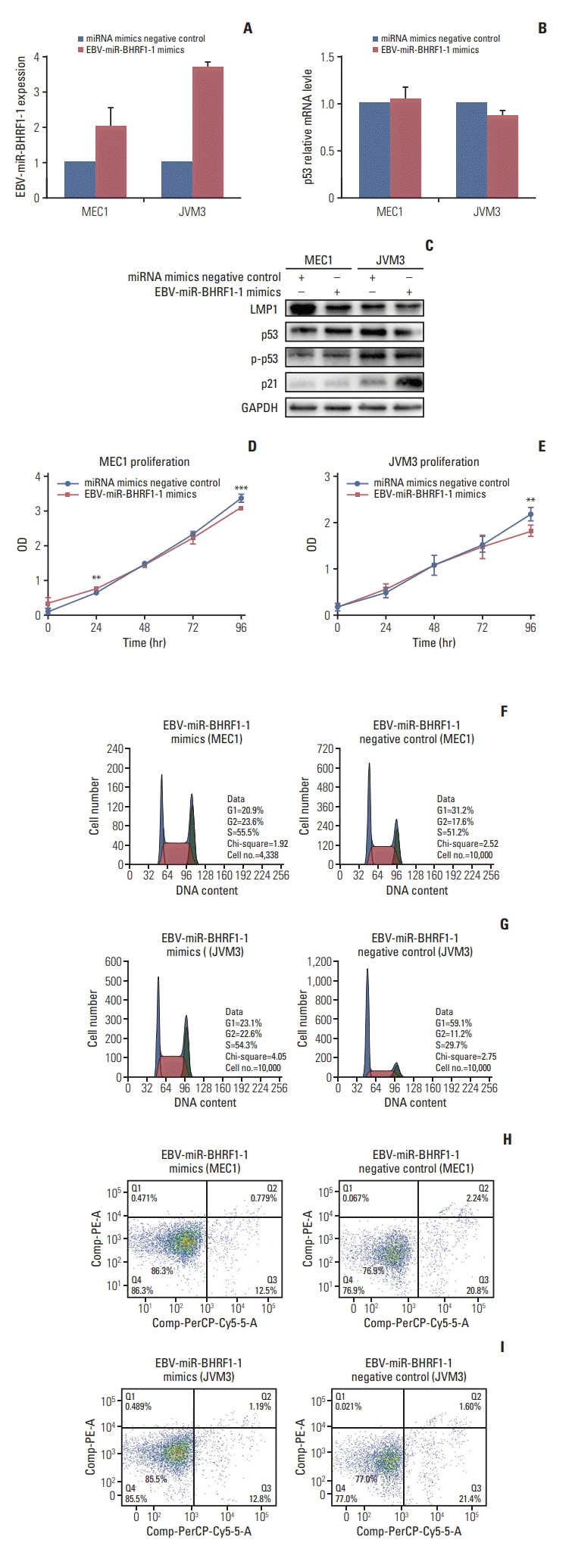
7. EBV-miR-BHRF1-1 mimics down-regulate p53 expression and affected CLL cell functions in MEC1 and JVM3
We then explored the biological effects of overexpression of EBV-miR-BHRF1-1 on cell lines of MEC1 and JVM3 and whether these effects may be mediated through decrease of p53 levels. We only used EBV-miR-BHRF1-1 mimic to increase the EBV-miR-BHRF1-1 levels in cell lines. qRT-PCR, WB and cell function tests were conducted after stably transfected. A significant increase of EBV-miR-BHRF1-1 expression level was observed in JVM3 (Fig. 5A). Although p53 mRNA level remained unchanged on EBV-miR-BHRF1-1 mimic transfection compared with NC (Fig. 5B), WB revealed down-expression of p53 and p21 protein (Fig. 5C). The CCK8 experiments were done to detect the proliferation ability of cell in 24, 48, 72, and 96 hours. Higher proliferation ability was observed in MEC1 and JVM3 cell lines transfected with EBV-miR-BHRF1-1 mimic compared with MEC1 and JVM3 cell lines transfected with miRNA NC at 96 hours (Fig. 5D and E). As shown in Fig. 5F-G, the G0/G1 phase was increased from 20.9% in MEC1 cells transfected with EBV-miR-BHRF1-1 mimic, to 31.2% on miRNA mimic NC (p < 0.05). Furthermore, the G0/G1 phase was increased from 23.1% in JVM3 cells transfected with EBV-miR-BHRF1-1 mimic, to 59.1% on miRNA mimic NC (p < 0.05). Apoptosis assay was performed to ascertain the biological effect of EBV-miR-BHRF1-1 mimics on the apoptosis of cell lines. A marked decrease in apoptosis was observed in the MEC1 and JVM3 cell lines transfected with EBV-miR-BHRF1-1 mimic (Fig. 5H and I).
Discussion
In this study, we show that high expression levels of EBV-BHRF1-1 in CLL patients were predictive of inferior TTT and OS. The predictive power of OS was retained after adjusted for all the prognostic factors in International Prognostic Index for patients with CLL [13]. We further explored the pathologic role of EBV in EBV-associated CLL and provided functional evidence that p53 was targeted by one of EBV miRNAs, EBV-miR-BHRF-1, which could affect the growth, proliferation, and apoptosis of EBV-positive CLL cells.
Thought the clues of EBV in CLL patients have been reported in several studies, we still did not know the latency type of EBV in EBV-positive CLL patients. Combined with the facts that LMP1 mRNA reported by Tarrand et al. [14] and EBV-miR-BHRF1-1 in our study have been detected in EBV-positive CLL patients, we hypothesized that EBV-positive CLL might be stage III type of latency, in which all latent genes, including LMP1, LMP2A, LMP2B, EBNA2, and EBNAs3-6 are all expressed and both EBV miRNAs of BHRF1 and BART clusters. Therefore, similar signaling pathways abnormities such as nuclear factor кB and p53 caused by EBV latency genes and EBV miRNAs might be also presented in EBV-positive CLL patients. In our study, we found that EBV-DNA positive CLL patients expressed higher levels of EBV-BHRF1-1 than EBV-DNA negative ones in CD19+ CD5+ B cells while the expression levels of EBV-DNA negative ones were similar to the normal healthy patients which were similar to the result reported by Visco et al. [7]. It can be easily explained by the infection of lytic infection in these CLL patients. Similar to our previous reports [6], high levels of EBV-BHRF1-1 were associated with inferior survival in CLL patients. Furthermore, high levels of EBV-BHRF1-1 were associated with elevated LDH levels, IGHV unmutated status and p53 aberration, suggesting a possible role of EBV-BHRF1-1 in the events that lead to CLL progression.
The study reported by Li et al. [11] has described p53 as a functionally relevant target of EBV-BHRF1-1 to potentiate lytic replication of EBV in nasopharyngeal carcinoma. Furthermore, several studies have demonstrated that lymphomas driven by EBV are dependent upon MDM2 which can further regulate p53 gene [15,16]. We also found that higher expressions of EBV-BHRF1-1 were associated with lower p53 and p53 protein which can reflect the functional role of p53 gene in our CLL patients. We further demonstrated for the first time that p53 is a potential miRNA target with likely pathogenic consequences in two mostly used two CLL cell lines (MEC1 and JVM3). Both MEC1 (with p53 mutation) and JVM3 (without p53 mutation) are EBV-positive cell lines with relatively higher EBV-BHRF1-1 levels. Therefore, MEC1 and JVM3 might be the optimal cell lines for us to validate the fact that p53 is indeed a cellular target for EBV-miR-BHRF1-1. In this study, we found that even a modest increase in p53 on treatment of MEC1 and JVM3 cells with miR-BHRF1-1 inhibitor is sufficient to alter cell cycle, inducing CLL cell apoptosis and inhibit the CLL cell proliferation. Similar results were also found by overexpression of EBV-miR-BHRF1-1 in MEC1 and JVM3 cells. From these results, we found that EBV-miR-BHRF1-1 inhibitor can affect the CLL cells in both p53 mutation and no p53 mutation cell lines. Therefore, EBV-miR-BHRF1-1 inhibitor might be a potential targeted drug in a small cohort of EBV-positive CLL patients with the high-risk cytogenetics feature of p53 aberration whose inferior prognosis cannot be overcome by current treatment strategies such as the conventional fludarabine based chemotherapy and newly molecular targeted drugs of BTK inhibitor and Bcl-2 inhibitors-based chemotherapy.
In conclusion, we presented evidence that p53 is a target of EBV-miR-BHRF1-1, which might be the underlying pathologic mechanisms for the inferior prognostic value for high expression levels of EBV-miR-BHRF1-1 in patients with CLL. The interaction between EBV-miR-BHRF1-1 and p53 may represent one of the many ways by which EBV-miR-BHRF1- 1 promotes EBV-positive lymphomagenesis of latency III type. Our results support the potential of EBV-miR-BHRF1- 1 as a therapeutic target for CLL patients with p53 gene aberrations.
 XML Download
XML Download