

 PDF
PDF Citation
Citation Print
Print


Introduction
BRCA1 is a tumor suppressor gene, and its mutation is significantly associated with hereditary breast cancer (BC) and ovarian cancer (OC) (HBOC) syndrome [1,2]. A variant of unknown significance (VUS) is a genetic variant that has been identified by genetic testing but of which the functional significance is not fully understood at this time [3]. Although considerable effort has been put into reclassification of VUSs, still, standardized guidelines have yet to be established.
The BRCA1 c.5339T>C, p.Leu1780Pro (L1780P) variant was considered a VUS until previous studies revealed that it is associated with HBOC syndrome [4-6]. Three studies from Korea suggested that this variant showed highly suspected features of a pathogenic BRCA1 mutation. However, all the previous studies included fewer than 20 patients; thus, there is a degree of uncertainty about the pathogenicity of the L1780P mutation that has remained. Furthermore, interestingly, reports of this variant are confined to the Korean population, and this variant has not been identified in Western countries [7-10].
We evaluated the clinicopathologic features of the L1780P variant in patients with BC using retrospective multicenter data from Korea to verify L1780P as a pathogenic BRCA1 mutation. We also compared the characteristics related to HBOC syndrome to identify HBOC-related characteristics between the L1780P variant and other locations of BRCA1/2 mutations.
Materials and Methods
1. Patients
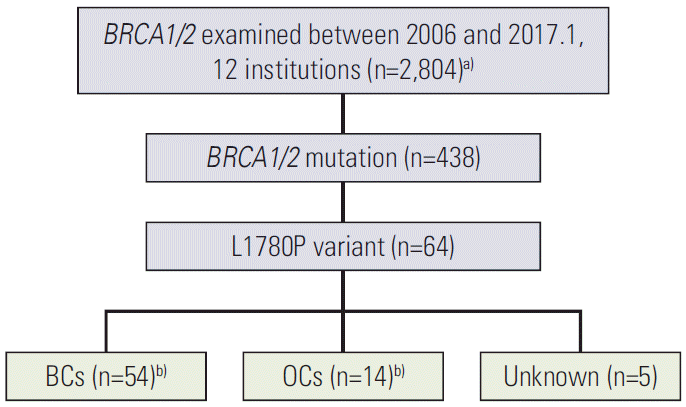
Twelve institutions initially agreed to participate in this study. Ten of them subsequently provided data from patients with L1780P (S1 A and S1B Fig.). Two institutions agreed to participate in the study but could not identify patients with the L1780P variant. One institute did not provide information on the number of BRCA1/2 mutations. There was insufficient clinical information for five patients with L1780P. Finally, a total of 64 patients with L1780P and BC and/or OC from the 10 institutions was enrolled in this retrospective study (Fig. 1).
The clinicopathologic data and information were reviewed. The clinicopathologic data included parity, marital status, age at diagnosis, family history (FHx), and personal history (PHx) of BC and/or OC, bilaterality of BC, pathologic stage according to the seventh American Joint Committee on Cancer classification, estrogen receptor (ER), and progesterone receptor (PR) status, human epidermal growth factor-2 (HER2) status, nuclear grade, Ki-67 status, type(s) of operation, type(s) of adjuvant treatment, risk-reducing procedures, sampling date, genetic testing method(s), and tumor subtype(s). We compared the HBOC-related characteristics in the patients with the L1780P variant to those of BC patients from the Korean Hereditary Breast Cancer (KOHBRA) study. Detailed information of enrolled patients in the KOHBRA study has been previously described [11]. The clinical outcomes were analyzed considering the following variables: recurrence, metachronous contralateral BC, and mortality. Personal identifiers for all patients were removed prior to analysis.
2. Diagnostic method
The BRCA1 L1780P variant was diagnosed using Sanger sequencing (62 patients), next-generation sequencing (one patient), conformation sensitive gel electrophoresis (one patient), and denaturing high-performance liquid chromatography (one patient). Detailed information of the diagnostic methods in the KOHBRA study was described previously [11].
3. Statistics
Patient characteristics were compared using independent t test for continuous variables and chi-square or Fisher exact test for categorical variables. Values are reported as mean±standard deviation or median with range. Survival curves were plotted using the Kaplan-Meier method. Recurrencefree survival (RFS) rate was calculated from the date of diagnosis or operation to the date of recurrence. Event-free survival (EFS) rate was calculated from the date of diagnosis or operation to the date of any event related to BC and/or OC. RFS was defined as any recurrence related to BC (e.g., local recurrence, distant metastasis, contralateral BC, and death due to BC). EFS was defined as any recurrence related to BC and/or OC (local recurrence, distant metastasis, contralateral BC, OC, and death by any cause). All tests were two-sided. A p-value of < 0.05 was considered statistically significant. The SAS ver. 9.4 (SAS Institute Inc., Cary, NC) and R3.2.1 (http://www.R-project.org; R Foundation for Statistical Computing, Vienna, Austria) software packages were used for statistical analyses.
4. Ethical statement
The World Medical Association Declaration of Helsinki on medical research protocols and ethics was adhered to throughout the procedures of this study. The Institutional Review Board (IRB) reviewed and approved this study (IRB number of National Cancer Center: 2017-04021, Kyungpook National University Hospital: 2015-05-205, Samsung Medical Center: 2016-09-038, Seoul National University Hospital: 1702-063831, St. Mary's Hospital: KC19REDI0387, Yonsei University Severance Hospital: 4-2016-1116). The need for informed consent was waived because of the low risk posed by this investigation. The authors have declared that no competing interests exist.
Results
1. Prevalence
A schematic diagram of patient selection is shown in Fig. 1. Overall 2,804 BRCA1/2 mutation tests were performed in patients with BC and/or OC. Of those, 438 patients (15.6%) had a BRCA1/2 mutation and 64 of 438 patients (14.6%) had the L1780P variant. One thousand six hundred sixty-nine BC patients in KOHBRA, 102 BRCA1 (6.1%), 143 BRCA2 (8.6%), and two BRCA1/2 (0.1%) mutations were identified, while 1,442 patients (86.4%) did not show BRCA1/2 mutation.
2. Patient characteristics
The clinicopathologic and treatment characteristics of the 54 BC patients with the L1780P variant are summarized in Table 1. Only one patient (1.9%) showed a low histologic grade while 41 patients (75.9%) demonstrated an intermediate to high nuclear grade. More than two-thirds of patients showed ER and PR negativity (ER negativity: 42/54, 77.8% and PR negativity: 45/54, 83.3%). HER2 overexpression was present in only two patients (3.7%). Triple-negative breast cancer was the most common subtype (41/54, 75.9%).
3. Clinical features related to HBOC syndrome in the patient with L1780P compared with patients with BRCA1/2 mutation in KOHBRA
The characteristics related to HBOC syndrome in patients L1780P and those with BRCA1/2 mutation in KOHBRA are shown in Table 2. There was no significant difference in mean age of BC or age group of BC (≤ 40 and > 40 year groups) between cases with L1780P and those with BRCA1/2 mutation in KOHBRA (L1780P vs. BRCA1 vs. BRCA2, p > 0.999 and p=0.107, p=0.652 and p=0.144, respectively). Patients with L1780P were more likely to have FHx of BC or OC (48/54, 88.9%) than patients with BRCA1/2 mutation in KOHBRA (75.3%, 186/241, p < 0.001).
Patients with L1780P were more likely to have FHx of OC (24.1%) and PHx of OC (16.7%) than those with BRCA1/2 mutation in KOHBRA (p < 0.001 and p=0.001, p=0.003 and p=0.001, respectively). There was no significant difference in FHx of BC, bilateral BC, or number of relatives with BC between L1780P and BRCA1/2 mutation groups, but the L1780P variant group was more likely to have FHx of OC (24.1% vs. 19.6% vs. 11.2%, p < 0.001 and p < 0.001), and PHx of OC (16.7% vs. 2.9% vs. 1.3%, p=0.009 and p < 0.001). In addition, the L1780P variant group had more number of relatives with BC than BRCA1/2 mutation of KOHBRA (all, p < 0.001). There was no significant difference in the number of relatives with OC between L1780P BRCA1/2 mutation of KOHBRA (p=0.351 and p=0.166, respectively). There was no significant difference in the median age of OC onset (p=0.615 and p=0.800, respectively).
4. Clinical outcomes related to HBOC syndrome
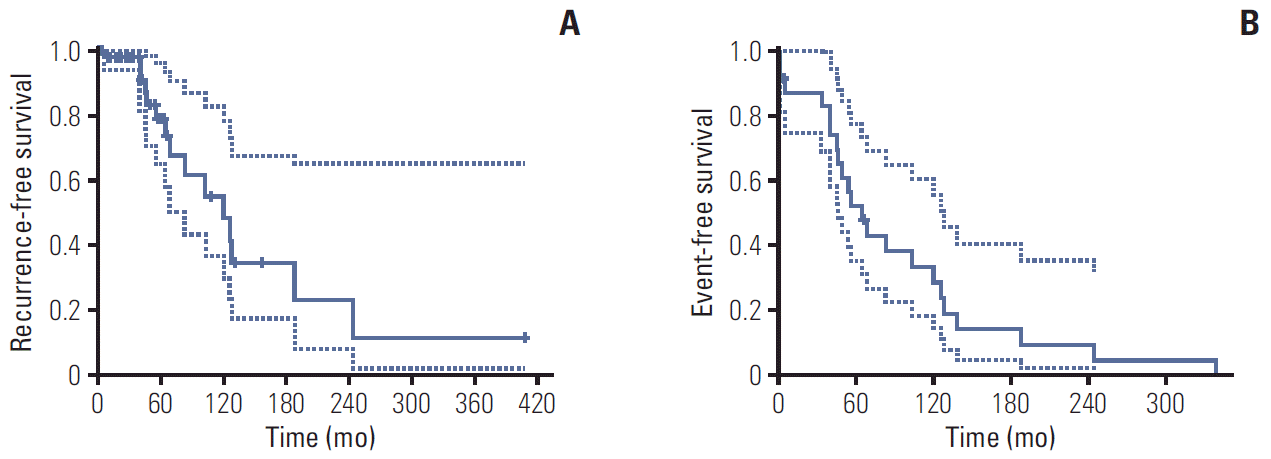
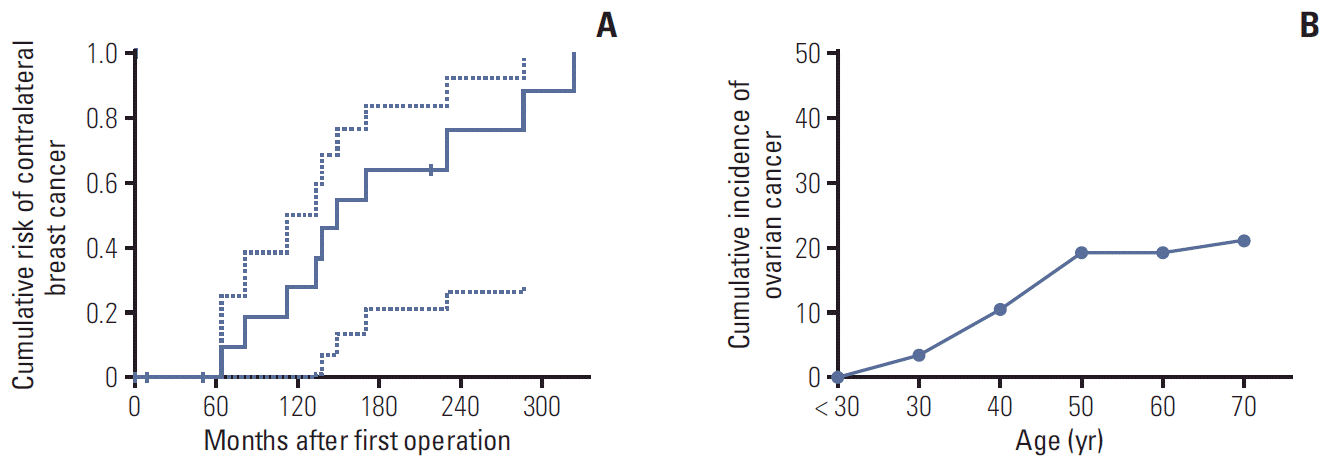
RFS and EFS curves are shown in Fig. 2. The 5- and 10-year RFS rates of patients with L1780P were 78.9% and 48.3%, respectively (Fig. 2A), and the 5- and 10-year EFS rates of patients with L1780P were 52.4% and 28.8 (Fig. 2B). Thirteen BC patients demonstrated bilateral breast cancer. The cumulative risk of contralateral BC at 10 years after diagnosis was 31.9%, while the cumulative risk of OC at 50 years of age was 20.0% (Fig. 3A and B).
Discussion
This study clearly demonstrated the clinicopathologic features of patients with L1780P mutation using data collected from 10 institutions in Korea and compared to other locations of BRCA1/2 mutation of KOHBRA. To our knowledge, this study is the largest study of patients with L1780P variants and provides clear evidence that patients with L1780P have similar clinicopathologic features to those of patients with a pathogenic BRCA1 mutation. Patients with the L1780P variant were more likely to have more frequent FHx and PHx of OC than patients with other BRCA1/2 mutations in the KOHBRA study.
Some pathogenic variants occur with high frequency because of a “founder effect” in particular racial/ethnic groups. Several founder mutations have been identified in specific populations, including Ashkenazi Jews [12-15]. The three well-known founder mutations in the Ashkenazi Jew population, BRCA1-185delAG, BRCA1-5382insC, and BRCA2-6174delT pathogenic variants, account for about 90% of all BRCA1/2 mutations in that population [16]. Interestingly, the L1780P variant showed high frequency only in the Korean population (Table 3). Previous research highlighted 33 patients with L1780P variant, with the prevalence of the L1780P variant of BC and OC being 26 of 2,147 (1.2%) and 11 of 436 (2.5%) and the proportions of the L1780P variant of BRCA1/2 and BRCA1 being 33 of 470 (7.0%) and 255 (12.9%), respectively [4-6,17]. In this study, 64 of 458 patients with a BRCA1/2 mutation (12.8%) showed the L1780P variant. This suggests that L1780P is a common mutation in Korean patients with HBOC syndrome. In this study, among the seven patients (13.5%) with family members with L1780P, the FHx of six patients (11.1%) showed co-segregation patterns of BC and/ or OC. The L1780P variant has been introduced as a likely pathogenic variant in recent years; however, segregation data from family members and haplotype analysis are not available, and such investigations are necessary to confirm whether L1780P is a founder mutation.
There are several reasons why a frequent and likely pathogenic variant (L1780P) had been classified as a VUS. One reason for its classification as a VUS may be the rare prevalence in Western countries. There was only one carrier who was of Asian ethnicity reported in the BIC database (http://research.nhgri.nih.gov/bic/). Since the KOHBRA study was started in 2007, the number of patients undergoing BRCA1/2 genetic testing has increased rapidly due to coverage of BRCA1/2 genetic testing by the National Health Insurance system of the South Korean government after May 2012 and the onset of the “Angelina Jolie effect” in May 2013 [11,18,19]. However, reclassification of VUSs in the Korean population is still not adequately performed due to limited data compared with Western public databases such as BIC or ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/). VUSs continue to be uncovered as BRCA1/2 pathogenic variants, especially in developing countries. Of note, Korean clinicians asked Myrid about the possibility of L1780P as a pathogenic variant and categorized L1780P as a VUS according to previous functional studies [20,21]. Several years later, they amended that L1780P has been reclassified to a suspected deleterious mutation based on a recent functional study and clinical report [4-6]. Even with many efforts to reduce VUSs, there is still a possibility that more pathogenic variants are classified as VUSs in the Korean population [22,23]. Thus, we should collect more data using nationwide or global databases to reduce the rates of VUS designation.
Rebbeck et al. [24] reported that BC and OC risks varied by type and location of BRCA1/2 mutation, and that risk assessment and cancer prevention decision making for carriers should be approached based on the location of BRCA1/2 mutation. The author demonstrated three breast cancer cluster regions (BCCRs) (c.179 to c.505, c.4328 to c.4945, and c.5261 to c.5563) and one ovarian cancer cluster region in BRCA1 mutation. Interestingly, L1780P is located in the BCCR but showed more likely to have OC compare to data of KOHBRA. Recently, Park et al. [25] demonstrated that patients with L1780P showed a relative risk of BC similar to that of carriers with other known deleterious mutations located in the BRCT domains of BRCA1. In addition, age at diagnosis of BC in patients with L1780P (median, 38.4 years; 95% confidence interval [CI], 33.9 to 42.9) was significantly earlier than that of carriers with mutations outside of the putative functional domain regions (median, 51.0 years; 95% CI, 45.0 to 57.0 years; p=0.017). In this study, the mean age at diagnosis of BC in patients with L1780P showed no significant difference compared with that of patients with BRCA1 mutation in KOHBRA. Further investigations are needed to validate the location of BRCA1/2 mutation and the risk of cancer prevalence in Korea as well as the other parts of the world, especially in northeastern Asian countries.
There are some limitations in the present study, including its retrospective design and loss of data collection. However, a prospective cohort study on this topic may be nearly impossible and is not a suitable design for research on rare mutations in a specific population. Furthermore, to the best of our knowledge, the current study presents the largest number of patients with the L1780P variants to date and demonstrates clear clinicopathologic evidence that the L1780P variant is a pathogenic variant of BRCA1.
In conclusion, patients with L1780P variant showed similar clinicopathologic features to patients with BRCA1 pathogenic mutation, so that L1780P variant in BRCA1 should be considered as pathogenic BRCA1 mutation. RRSO should be highly recommended for women with the L1780P variant.
 XML Download
XML Download