

 PDF
PDF Citation
Citation Print
Print


Introduction
Oncolytic viruses are an emerging class of cancer therapeutic agents that exert antitumor effects generally through tumor-specific cell lysis paired with immune stimulation [1]. The development of a new oncolytic virus usually involves genetic engineering of a backbone virus. Viruses that are frequently chosen for genetic engineering include vaccinia virus, adenovirus, and herpes simplex virus. Vaccinia virus has many ideal characteristics that allow for development as an oncolytic virus, such as its relatively large genome as an optimal platform for gene editing, long usage history, and well-defined safety profile [2]. Techniques that are commonly used for genetic engineering include deletion and insertion of specific genes within a virus genome. Forinstance, interruption of the virus thymidine kinase-encoding TK gene through inserting other transgenes has been applied in the development of several oncolytic viruses [3,4]. The rationale is twofold: first, a virus without a functional TK gene can only amplify extensively inside cells with high level of thymidine kinase, which is a distinct feature of many cancer cells [5]; second, an inserted transgene may introduce new antitumor mechanisms to the oncolytic virus.
To this end, we designed and engineered SJ-185, a novel oncolytic vaccinia virus, through incorporating two human transgenes, the IFNB1 and CES2, into the TK gene region of a Western Reserve (WR) strain vaccinia virus backbone. Interferon β1 (IFN-β1), the product of the IFNB1 gene, is a cytokine with indisputable antitumor effects [6,7]. However, it has a very short half-life and its antitumor effects require high tissue concentration [8]. We hypothesized that the insertion of IFNB1 transgene may enable SJ-815 more continuously produce high-levels of IFN-β1 within tumor tissue, which is difficult to achieve using conventional drug delivery systems. In addition, low level of IFN-β1 expressed in normal tissue may help eliminate SJ-815 and reduce potential side effects [9]. Unlike the IFNB1 gene, the CES2 gene does not produce molecules that kill cancer cells directly. We hypothesized that this transgene of SJ-815 may increase tumor-specific expression of carboxylesterase 2, and subsequently, increase the tumor’s capacity to metabolize irinotecan. Irinotecan is one of the most widely used chemotherapeutic agents and has been included in the World Health Organization (WHO) Model List of Essential Medicines [10]. It is most well-known as a treatment for colon cancer, but recently it has been used in combination with other drugs (such as folinic acid, fluorouracil, and oxaliplatin) for treatment of metastatic pancreatic cancer [11], which received approval in countries such as the United States, and South Korea, and the European Union. Irinotecan is a prodrug that needs to be activated into SN-38 in the human body, namely by carboxylesterase 2 [12]. Carboxylesterase 2 shows great variability among individuals [12,13], and higher carboxylesterase 2 activity in tumor tissue predicts better survival in irinotecan-treated cancer patients [14]; therefore, it is critical to improve irinotecan metabolism for patients with low carboxylesterase 2 activity. We hypothesized that tumor-specific carboxylesterase 2 enhancement by SJ-815 may be more advantageous than a systemic enhancement of carboxylesterase 2 because the latter may increase the risk of SN-38 induced intestinal toxicity [15].
The life cycle of vaccinia virus consists of four different forms: intracellular mature virus, intracellular enveloped virus (IEV), cell associated virus, and extracellular enveloped virus (EEV) [16]. EEV is the ultimate mature form of vaccinia virus, and it plays an important role in the long-range viral dissemination (both in vitro and in vivo), a function that cannot be fulfilled by the other forms [16]. In this study, considering the insertion of the human IFNB1 gene may result in decreased viral dissemination [17], WI, a TK deleted Western Reserve vaccinia virus with a point mutation (451A>G) in the A34R gene that results in improved viral dissemination was used for genetic engineering [18]. A previous study shows that this mutation can cause over a 10-fold increase in EEV formation [18].
Therefore, the objectives of this study were to characterize SJ-815 in terms of its in vitro viral replication and dissemination, tumor cytotoxicity, and in vivo irinotecan metabolism; as well as to evaluate its in vivo antitumor effects, particularly when it is combined with irinotecan.
Materials and Methods
1. Engineering of SJ-815
The backbone virus, named WI, used in this study was WR A34R-451A>G TK-, kindly provided by Dr. Stephen H. Thorne (University of Pittsburgh, Pittsburgh, PA) [19]. Two human transgenes (the IFNB1 and CES2 genes) were inserted into the TK gene region of the WI virus through homologous recombination, using the pSC65 vector that carries both genes (Genewiz Inc., South Plainfield, NJ). The CES2 (14 kbp; NCBI gene ID: 8824) and IFNB1 (1.1 kbp; NBCI gene ID: 3456) genes were placed under the control of p7.5 (vaccinia early/late promotor) and pSE/L (vaccinia synthetic early/late promotor), respectively. The successfully engineered virus, named SJ-815, was WR A34R-451A>G TK– CES2+ IFNB1+. Pure plaques of SJ-815 were obtained from the 143B cell line (human osteosarcoma, American Type Culture Collection [ATCC], Manassas, VA), which is a TK gene negative cell line maintained under selective pressure of 5-bromo-2’-deoxyuridine (Sigma-Aldrich, St. Louis, MO) and tested for transgene expression described in next paragraph. Selected plaques were amplified in the human cervical cancer cell lines HeLa and HeLa S3 cells (ATCC), which were cultured with Dulbecco’s modified Eagle medium (DMEM; Hyclone, Westborough, MA) supplemented with 2.5% fetal bovine serum (FBS; Hyclone). The cells and medium were then harvested and homogenized. The homogenized samples were purified through sucrose cushion centrifugation consisting of 36% sucrose in 10 mM Tris, pH 9.0, and purified virus was re-suspended in the final stock buffer, 1 mM Tris, pH 9.0. The mouse version SJ-815, named mSJ-815, which has the murine IFNB1 gene (1.0 kbp; NCBI gene ID: 15977) instead of the human IFNB1 gene, was made with a similar method as described above.
2. Confirmation of CES2 and IFNB1 insertion and expression
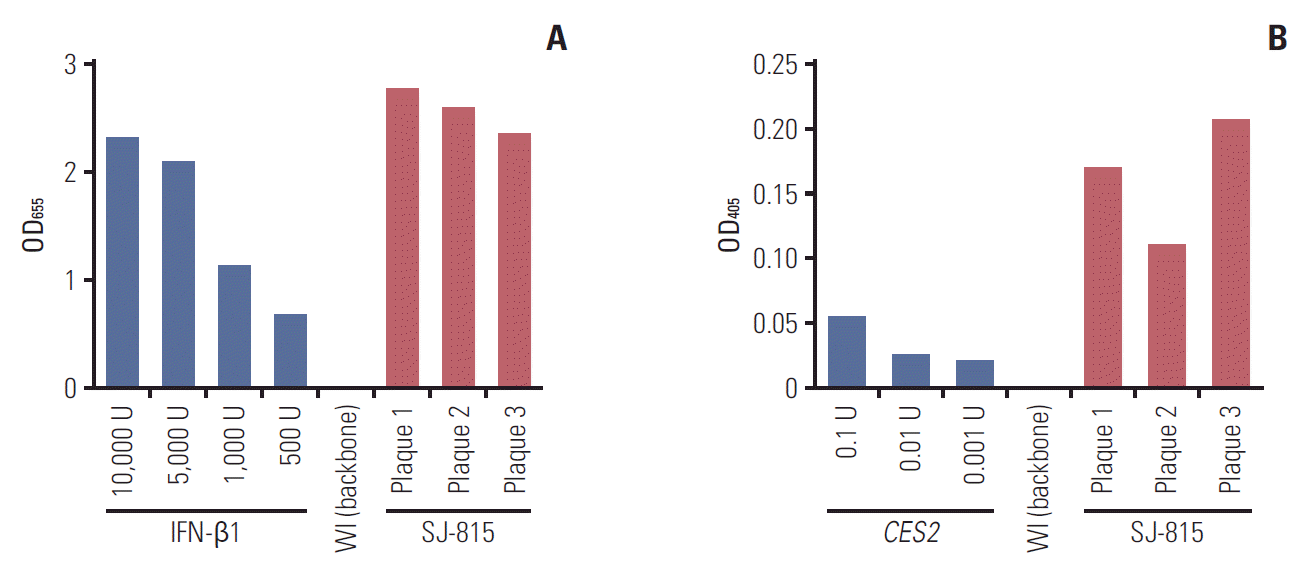
Polymerase chain reaction (PCR) analyses were conducted to confirm the insertion of the two transgenes. The expression of IFNB1 was determined using the HEK-Blue IFN-α/β cells (cat No. hkb-ifnab, InvivoGene, San Diego, CA). These cells were engineered to express the ISGF3 signaling pathway which is activated specifically by type I interferon. The product of this pathway, secreted embryonic alkaline phosphatase, was then measured spectrophotometrically at 655 nm to indicate the IFN-β1 activity. The protein expression of CES2 was determined using a p-nitrophenyl acetate (PNPA; 10 mM in dimethylsulfoxide) assay. PNPA is a carboxylesterase specific substrate; the production of its metabolite p-nitrophenol was measured spectrophotometrically at 405 nm to indicate the carboxylesterase 2 activity. One unit (U) of esterase activity was defined as the amount of enzyme releasing 1 μmol p-nitrophenol per min under assay conditions.
3. Measurement of viral replication
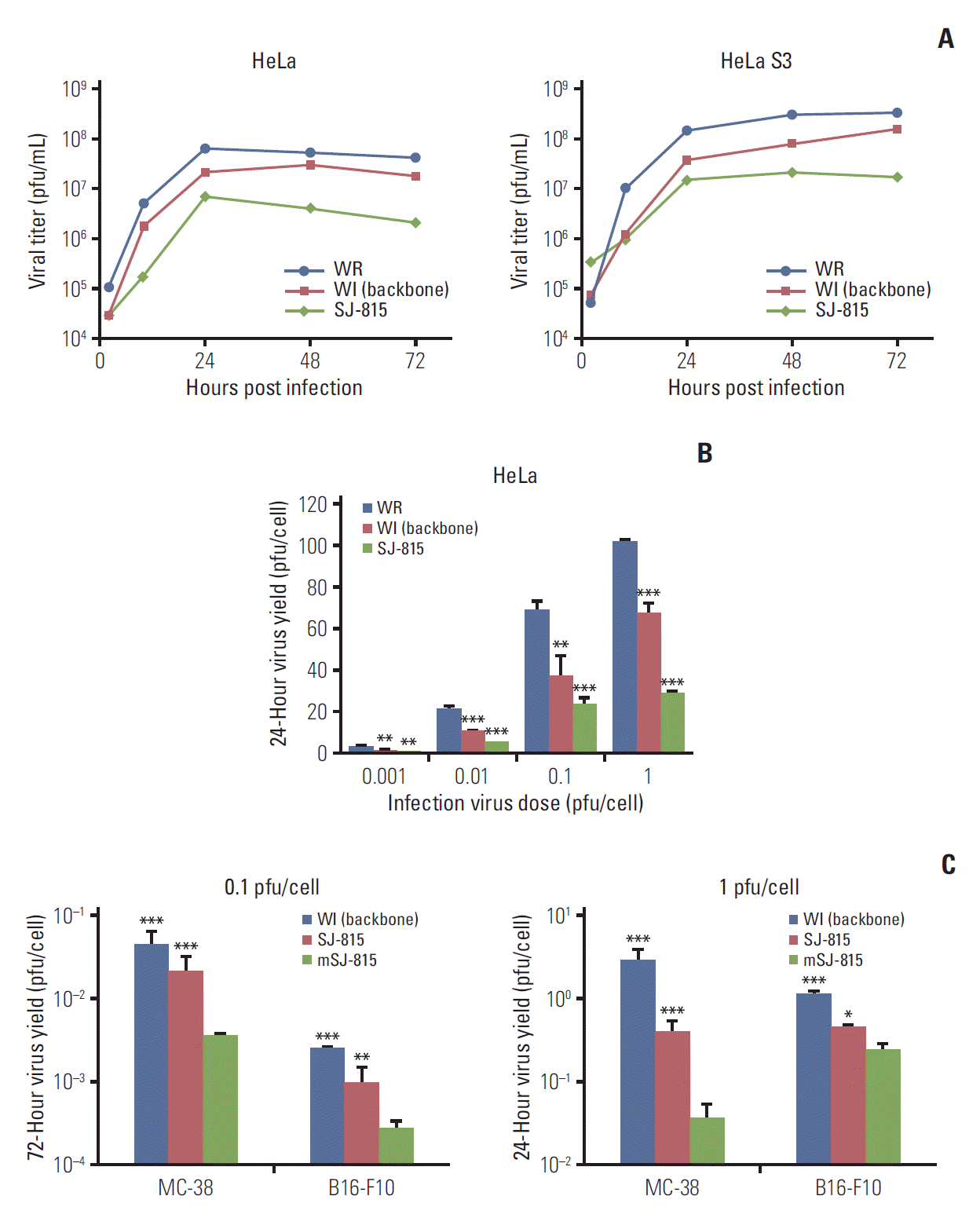
To analyze time-dependent virus growth, HeLa and HeLa S3 cells (ATCC) were seeded in 9-well tissue culture plates, and then infected with virus (WR, WI or SJ-815, 3 pfu/cell). After being incubated at 37°C in 5% CO2 for 2 hours, the supernatant was aspirated and the cells were washed twice with DMEM containing 2.5% FBS. The infected cells were overlaid with DMEM containing 2.5% FBS and incubated at 37°C in 5% CO2 for 2, 24, 48, or 72 hours, and the stocks were lysed by three freeze-thaw cycles and sonication. Vial titers were determined using a plaque assay: U-2 OS cells seeded on 6-well plate were infected by serially diluted virus samples and incubated at 37°C in 5% CO2 for 2 hours. The supernatant was aspirated and the cells were washed twice with DMEM containing 2.5% FBS, the plates were then overlaid by semi-solid carboxymethyl cellulose (CMC) medium (3% CMC solution diluted in same volume of 2×DMEM containing 4% FBS) and incubated for 72 hours at 37°C in 5% CO2 for 72 hours. The CMC overlay was then aspirated and the wells were stained with 2 mL of 0.1% crystal violet solution.
A similar study procedure was used to analyze the following: (1) virus dose-dependent virus yield, except that only HeLa cells were used, and four virus doses were chosen for virus infection (0.001, 0.01, 0.1, and 1 pfu/cell), and the incubation time was 24 hours; and (2) viral replication of WI, SJ-815, and mSJ-815 in MC-38 and B16-F10 mouse cancer cells (Korean Cell Line Bank, Seoul, Korea).
4. Evaluation of viral dissemination
To evaluate viral dissemination, comet assays were used to measure EEV formation. U-2 OS cells (ATCC) were infected with the virus (WR, WI, or SJ-815; 30 pfu/plate) and incubated at 37°C in 5% CO2 for 2 hours. The supernatant was then aspirated and the cells were washed twice with DMEM containing 2.5% FBS. The infected cells were overlaid with one of the following: DMEM containing 2.5% FBS, recombinant human IFN-β1 expressed in CHO cells (Sigma-Aldrich), or virus supernatants from other viruses (described below). All were incubated at 37°C in 5% CO2 at an angle of 10° for 72 hours. The virus supernatants were collected after the U-2 OS cells were infected with the viruses (WR, WI, or SJ-815) for 16 hours, and filtered with 0.22-μm filter (Merck Millipore, Billerica, MA) three times and stored at 4°C until use.
5. Evaluation of in vitro cytotoxicity of SJ-815
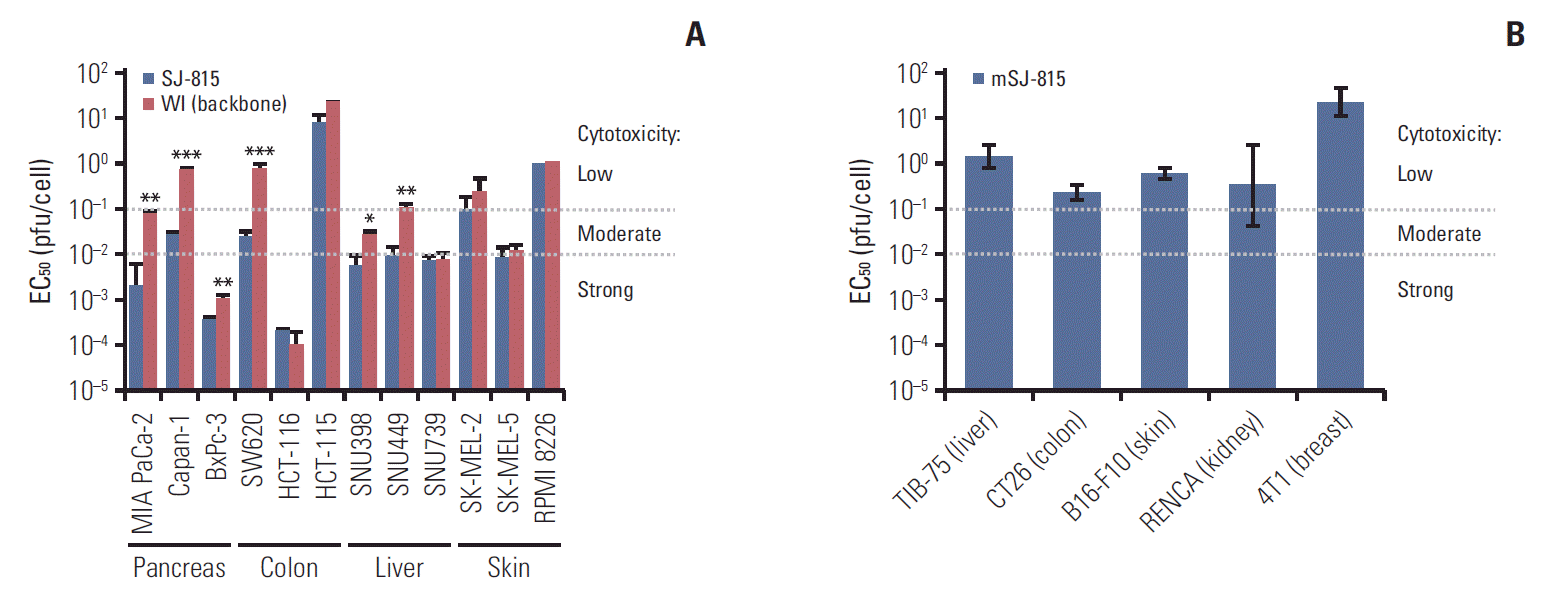
Twelve human cancer cell lines, three each for pancreas (MIA PaCa-2, Capan-1, and BxPc-3; ATCC), colon (SW620, HCT-115, and HCT-115; ATCC), liver (SNU398, SNU449, and SNU739; Korean Cell Line Bank), and skin (SK-MEL-2, SK-MEL-5, and RPMI8226; ATCC) cancers, and five murine cancer cell lines (ATCC), one each for liver (TIB-75), colon (CT26), skin (B16-F10), kidney (RENCA), and breast (4T1) cancers, were seeded at a concentration of 1×104 cells/well in 96-well plates containing 100 μL growth media per well, and incubated at 37°C in 5% CO2 for 24 hours cells were infected with 100 to 0.0001 pfu/cell (10-fold dilution each time) of SJ-815 or WI. The cell viability was assessed 48 hour post-infection using cell counting kit-8 (CCK-8; Dojindo, Kumamoto, Japan).
6. Evaluation of antitumor efficacy in xenograft mouse model
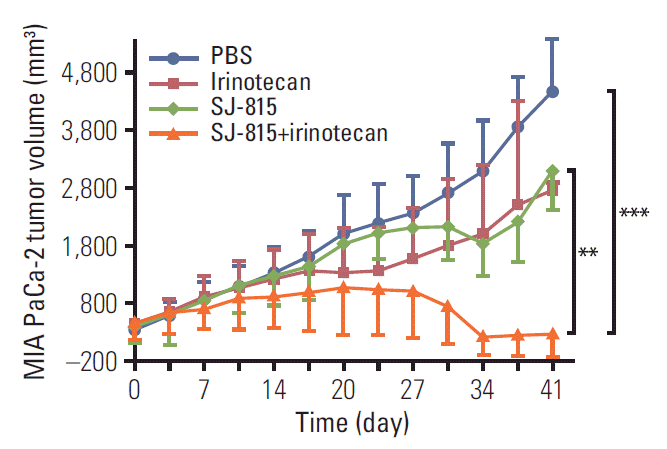
Female BALB/c nude mice (OrientBio, Seongnam, Korea) were injected subcutaneously with MIA PaCa-2 cells on the right flank and developed human pancreatic cancer. Once tumors reached a volume of 400 mm3, mice were randomized on day 0 into four treatment groups (n=3 mice/group): phosphate-buffered saline (PBS), SJ-815 (1×106 pfu; intratumoral; day 7 and 14), irinotecan (25 mg/kg; intravenous; day 3, 10, and 17), and SJ-815 (1×106 pfu; intratumoral; day 7 and 14)+irinotecan (25 mg/kg; intravenous; day 3, 10, and 17). Tumor measurements were performed with calipers twice per week and on sacrifice day (day 41).
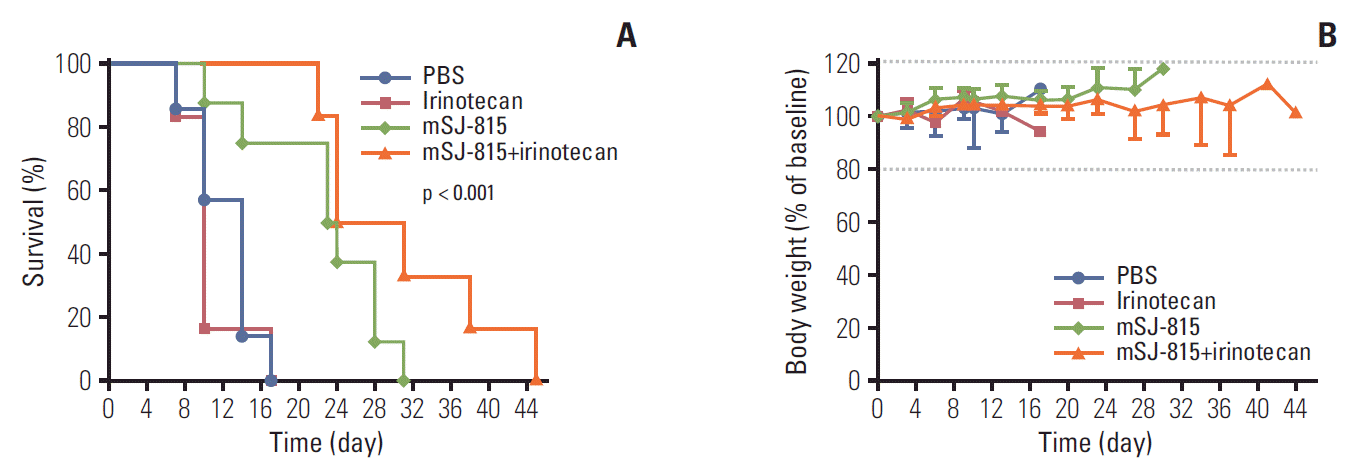
7. Evaluation of survival in syngeneic mouse model
Male C57BL/6 syngeneic mice (OrientBio) were injected subcutaneously with B16-F10 cells (1×105 cells/mouse) on the right flank and developed mouse melanoma. Once tumors reached a volume of 50-100 mm3, mice were randomized into four treatment groups: PBS (n=8), mSJ-815 (n=8; 3×107 pfu; intratumoral; day 7 and 14), irinotecan (n=6; 25 mg/kg; intravenous; day 3, 10, and 17), and mSJ-815 (n=6; 3×107 pfu; intratumoral; day 7 and 14)+irinotecan (25 mg/kg; intravenous; day 3, 10, and 17). Survival was recorded and body weight was measured twice a week.
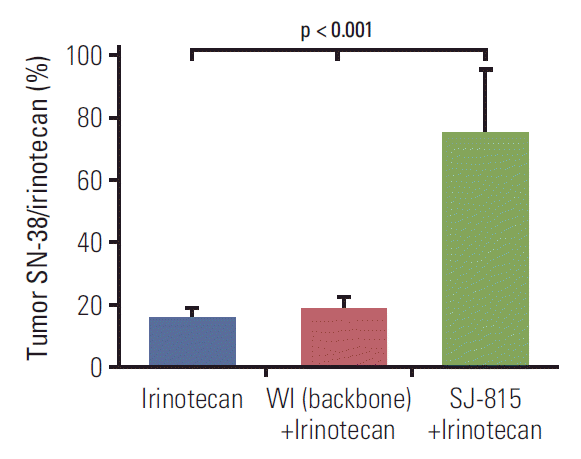
8. Measurement of irinotecan metabolism in vivo
Male C57BL/6 syngeneic mice were implanted subcutaneously with B16-F10 mouse melanoma cells (4×106 cells/mouse). Once tumors reached a volume of 100-200 mm3, mice were randomized into four treatment groups: PBS (n=5), irinotecan (n=5; 25 mg/kg; intravenous; day 3), WI (n=5; 1×107 pfu; intravenous; day 0)+irinotecan (25 mg/kg; intravenous; day 3), and mSJ-815 (n=5; 1×107 pfu; intravenous; day 0)+irinotecan (25 mg/kg; intravenous; day 3). All mice were sacrificed on day 7 and tumors were harvested.
The amount of irinotecan and SN-38 in tumor was measured as follows: 1 g of tumor was suspended in 3 mL of Hank’s Balanced Salt Solution without calcium and magnesium. Tissue suspension was homogenized 3 times (20 seconds each, with 1-minute intervals) and incubated in a water bath for 2 hours at 55°C after adding 15 μL of 20 μg/μL protease K. The tissues were homogenized 3 times again (20 seconds each time, with 1-minute intervals), and then vortexed for 5 minutes after 1 mL of 100% acetonitrile was added to each sample. Tissue suspension was centrifuged for 10 minutes at 4,500 rpm and supernatant was recovered and centrifuged again for 5 minutes at 13,000 rpm. The supernatant was then dilu-ted with 1.2 mL of diethyl ether: ethyl acetate (v:v=2:1) mixture and vortexed for 10 minutes. The supernatant was isolated again after centrifugation for 5 minutes at 13,000 rpm and dried using speed vac (SAVANT SC201A, Thermo Scientific, Waltham, MA) for 1 to 1.5 hours at 45°C. The final pellet was re-suspended in 100 μL of 100% methanol, vortexed and used for liquid chromatography–mass spectrometry analysis [20].
9. Statistical analysis
The in vitro experiments were conducted in duplicate or triplicate, and data were presented as mean±standard deviation (SD). All statistical analyses were conducted with the Prism software ver. 6 (GraphPad Software, La Jolla, CA). Data comparisons were done using t tests, paired t tests, or ANOVA tests. EC50 was calculated using nonlinear regression (the dose-response–inhibition module). The survival analysis was done using a log-rank test. A p-value < 0.05 was considered statistically significant.
10. Ethical statement
All animal care, handling and study procedures were in compliance with the Institutional Animal Care and Use Committee (IACUC) guidelines from the Korea Ministry of Food and Drug Safety. The animal study protocols were approved by the Pusan National University IACUC (PNU-2014-0673 and PNU-2015-0872) and performed at the Laboratory Animal Resource Center of Pusan National University, Korea.
Results
1. Confirmation of CES2 and IFNB1 insertion and expression in SJ-815
The insertion of the CES2 and IFNB1 transgene in the final selected virus plaques was confirmed by PCR analyses (data not shown). The in vitro function tests conducted in three virus samples demonstrated stable expression and activity of the two protein products IFN-β1 and carboxylesterase 2 (Fig. 1).
2. Decreased viral replication of SJ-815
At the infection virus dose of 3 pfu/cell, all three different viruses (WR, WI, and SJ-815) continued replicating for the first 24 hours in both HeLa and HeLa S3 cells and the replication reached plateau thereafter (Fig. 2A). The replication ability of the viruses in both cancer cells ranked in sequence: WR > WI > SJ-815. WR showed a replication around an order of magnitude higher than SJ-815. In addition, the differences in 24-hour replication ability between the three viruses became greater as the virus titer increased from 0.001 to 1 pfu/cell, as shown in HeLa cells (Fig. 2B). SJ-815 and WI showed relative lower replication in the mouse cancer cells than in the human cancer cells, and mSJ-815 demonstrated the lowest replication, which was significantly lower than SJ-815 and WI (Fig. 2C).
3. Decreased enveloped extracellular virus formation of SJ-815
Compared to the wild-type WR (Fig. 3A), the WI virus showed significantly increased formation of comet tails (Fig. 3B). Contrastingly, SJ-815 did not show any obvious formation of comet tails (Fig. 3C). In the supernatant exchange experiments, the comet tails formed in the WI virus culture were significantly reduced both by the media containing 1,000 U IFN-β1 and by the supernatant of SJ-815 culture; but were not changed by the supernatant of the WR virus culture (Fig. 3D). The comet tails formed in the SJ-815 culture were not changed by the supernatant of the WR virus culture, either (Fig. 3E).
4. In vitro tumor cytotoxicity of SJ-815 (mSJ-815)
Among the 12 human cancer cell lines, WI showed strong (EC50, < 0.01 pfu/cell) tumor cytotoxicity in two cell lines and moderate (EC50, 0.01-0.1 pfu/cell) tumor cytotoxicity in 3 cell lines; alternatively, SJ-815 showed strong tumor cytotoxicity in seven cell lines and moderate tumor cytotoxicity in three cell lines (Fig. 4A). SJ-815 showed over 10-fold higher tumor cytotoxicity than WI in four cell lines: MIA PaCa-2 (pancreas; 37 fold), Capan-1 (pancreas; 26 fold), SW620 (colon; 32 fold), and SNU449 (liver; 11 fold). However, mSJ-815 showed low (EC50, > 0.1 pfu/cell) tumor cytotoxicity in all five murine cancer cell lines tested (Fig. 4B).
5. In vivo antitumor efficacy and safety of SJ-815
In the MIA PaCa-2 (human pancreatic cancer)-bearing mouse model (Fig. 5), for the first 3 weeks, tumor growth was similar between the PBS, irinotecan, and SJ-815 groups. Mice in the irinotecan and the SJ-815 groups, from around day 20 and day 27, respectively, started to show slower tumor growth than that in the PBS group. On the sacrifice day (day 41), the average tumor sizes in the irinotecan (2,800 mm3) and SJ-815 groups (3,000 mm3) were smaller than in the PBS group (4,500 mm3) but the differences were not significant. The tumor growth in the SJ-815 plus irinotecan combination group was the slowest among all the groups, and tumor regression was observed after day 34: the tumor sizes shrank to below the baselines and remained stable (~200 mm3) until the sacrifice day (p < 0.01, vs. PBS group; p < 0.001, vs. SJ-815).
In the mSJ-815 insensitive B16-F10 (mouse melanoma)-bearing mouse model, the tumors developed very fast in the PBS and irinotecan groups (S1 Fig.), the Kaplan-Meier curves showed that irinotecan alone did not prolong the mice’s survival compared to PBS; the median survival for irinotecan and PBS groups were 10 and 14 days, respectively (Fig. 6A). Alternatively, in the mSJ-815 and mSJ-815+irinotecan combination groups, the tumor growth had remained relatively low for the first 3 weeks (S1 Fig.), mSJ-815 significantly increased the median survival to 24 days (hazard ratio [HR], 0.11; 95% confidence interval [CI], 0.02 to 0.50; p=0.013) and the mSJ-815+irinotecan combination group showed the longest median survival among all the groups (HR, 0.05; 95% CI, 0.01 to 0.19; p=0.001), which was 28 days (Fig. 6A).
The changes in body weight remained within 20% of the baseline throughout the study in all groups. No obvious toxicity was observed (Fig. 6B).
6. Conversion of irinotecan to SN-38 by SJ-815
In the B16-F10 tumor tissue obtained 7 days after intravenous virus injection, the SN-38/irinotecan ratios were similar between the irinotecan and WI+irinotecan combination groups, and both were less than 20%; in comparison, this ratio was over 70% and was significantly (p < 0.001) higher in the SJ-815+irinotecan group compared to the other groups (Fig. 7).
Discussion
This is the first report on SJ-815, a newly engineered oncolytic vaccinia virus which lacks a functional TK gene, and instead, expresses two human transgenes: IFNB1 and CES2. One encodes IFN-β1, a cytokine that is well-known for its antitumor and antiviral effects, and the other one encodes carboxylesterase 2, an enzyme that is critical for activation of irinotecan. SJ-815 demonstrates a few distinct characteristics in that it shows greatly reduced in vitro viral replication and dissemination, but significantly increased cytotoxicity against multiple tumor cell lines compared to its backbone virus, WI. Furthermore, SJ-815 shows potent antitumor effects in tumor-bearing mouse models, particularly when combined with irinotecan.
In this study, the length and width of comet tails in a comet assay were used to illustrate in vitro viral dissemination, because comet tails are formed specifically by the infectious and mature EEV [21]. In this study, the dissemination of wild-type WR virus was lower than WI virus, as expected; however, the dissemination of the wild-type WR virus can be significantly increased by the supernatant from the 16-hour WI culture, suggesting that a high level of EEV has been formed within 16 hours. Despite being engineered using the dissemination-enhanced WI virus, SJ-815 showed almost completely diminished comet tails. Interestingly, WI virus could not form comet tails if supernatant from the 16-hour SJ-815 culture was added. The diminished comet tails suggest that a considerable amount of IFN-β1 was persistently released into the supernatant within 72 hours, to the extent that the dissemination enhancing effect of the WI was completely offset. However, viral dissemination ability does not correlate with viral replication ability, as in this study, dissemination-enhanced WI virus demonstrated markedly lower replication than the wild-type WR virus in the two cell lines tested. The reduced viral replication is likely caused by the deletion of the TK gene [22,23]. Therefore, due to the deletion of the TK gene, as well as to the insertion of the IFNB1 gene, SJ-815 has both reduced viral replication and dissemination.
However, the tumor cytotoxicity of SJ-815 was superior to WI in nearly all (11 out of 12) human cancer cell lines tested, with considerably high potency (EC50 < 0.01 pfu/cell) in most (6 out of 9) gastrointestinal cancer cell lines. In the MIA PaCa2 (human pancreatic cancer)-bearing mouse model, both irinotecan and SJ-815 showed only moderate inhibition on tumor growth, suggesting that SJ-815 or irinotecan monotherapy is insufficient in controlling tumor growth. There are various causes for the in vitro and in vivo antitumor effects discrepancy, such as host antiviral immunity, tumor stromal barrier, in vitro-in vivo pharmacokinetic differences, etc. [24], but the combination use of SJ-815 plus irinotecan resulted in remarkable tumor regression (tumor size lower than baseline) in 5-6 weeks. It is worth mentioning that the treatments started with a much larger tumor size (around 400 mm3) than commonly used in mouse studies (50-200 mm3), to mimic a more advanced disease stage seen in most pancreatic cancer pati-ents. Similarly, in the highly aggressive B16-F10 (mouse melanoma)-bearing mouse model, the mSJ-815 monotherapy doubled the survival time compared to those treated by PBS or those by irinotecan only. It is also noteworthy that mouse cell lines in general are likely to be resistant to mSJ-815 replication, and therefore resistant to their cytotoxicity (EC50, > 0.1 pfu/cell). The combination use of mSJ-815 and irinotecan led to a striking 90% decrease in HR for survival. These animal studies suggest that combination use of irinotecan may largely potentiate antitumor effects of SJ-815/mSJ-815, even against SJ-815/mSJ-815 resistant tumors. Considering the complexity and heterogeneity of human cancer biology, as well as challenges associated with identifying biomarkers for tumor response prediction, any oncolytic virus monotherapy may only work for a limited number of cancer types, therefore, combination therapies of SJ-815 plus irinotecan may become a better treatment option.
The supernatant exchange experiments in the comet assays confirm that IFN-β1 is being continuously released from the SJ-815 infected cancer cells into tumor tissue. This is important because the antitumor effects of IFN-β1 come not only from direct anti-proliferative and anti-angiogenesis effects, but also from immune stimulation [6], e.g., activation of NK cells, maturation of myeloid cells into immunogenic antigen-presenting cells [25]. Both IFN-β1 and oncolytic viruses have been used as immune modulators to be combined with immune checkpoint inhibitors and have shown encouraging results [26,27]. Previously, a vesicular stomatitis virus expressing IFN-β1 was shown to be able to create a more optimal tumor microenvironment for immune checkpoint inhibition [28]. Therefore, it will be beneficial to conduct future studies to find out whether SJ-815 can significantly sensitize tumors to immune checkpoint inhibitors.
A previous study shows that virus-generated tumor expression of carboxylesterase 2 can potentiate the antitumor activity of irinotecan, regardless of whether the enzyme is expressed inside or outside the cells [29]. Therefore, it is suggested that the decreased EEV formation (which occurs outside of cancer cell) of SJ-815 may not greatly influence its ability to enhance irinotecan metabolism in tumor tissue. Previously reported SN-38/irinotecan ratios in tumor tissues following irinotecan intravenous injections vary greatly between tumor types, ranging from 10% to 50% after 8 hours [30-32]. In our study, the tumor SN-38/irinotecan ratio after intravenous administration of only irinotecan was below 20%, which was not changed with the combination intravenous administration of WI virus+irinotecan; however, this ratio was remarkably increased (by over 300%) by the combination intravenous administration of mSJ-815+irinotecan, suggesting that systemically injected SJ-815 can increase tumor expression of carboxylesterase 2 substantially. Since our previous study found no viral replication in normal tissues after systemic injections of a TK deleted vaccinia virus [33], it is likely that the SN-38/irinotecan ratio in the intestines would remain unchanged.
There are several limitations of this study: (1) Because SJ-815 did not show compromised antitumor effect in vitro compared to WI virus, for study simplicity, we did not measure the in vivo dissemination post SJ-815 treatment. (2) To minimize animal numbers, we did not include WR or WI control groups in the mouse studies; however, the small sample size, particularly in the MIA PaCa-2 study, may have prevented us from drawing a definite conclusion regarding the antitumor effects of SJ-815 and irinotecan as monotherapies. (3) Only body weight was measured to indicate the toxicity of SJ-815, more toxicity and safety information will be obtained in future studies from lab tests, imaging analyses, as well as other animal models (e.g., rabbits, monkeys). (4) Specific tumor microenvironment changes should be studied in order to better understand the mechanisms of action of SJ-815.
In conclusion, to our knowledge this is the first study that demonstrates the influences of human transgenes IFNB1 and CES2 on viral dissemination and irinotecan metabolism. And the novel oncolytic virus SJ-815 shows encouraging preclinical antitumor effects and no obvious toxicity. More studies are needed for safety and efficacy information.

 XML Download
XML Download