

 PDF
PDF Citation
Citation Print
Print


Introduction
Uterine cervical cancer is the fourth most commonly diagnosed cancer and the fourth leading cause of cancer death among women, with an estimated 570,000 new cases and 311,000 cervical cancer–related deaths reported worldwide in 2018. In low Human Development Index settings, it ranks second only to breast cancer in incidence and mortality [1]. The causal link between high-risk human papillomavirus (HPV) infection and cervical cancer, including squamous intraepithelial lesion (SIL), has been well established by molecular and epidemiologic studies [2]. However, HPV infection is self-limited and regresses within 1 year in most cases [3].
Cervical cancer screening is mainly based on Papanicolaou (Pap) tests, and Pap test–based screening has played an important role in reducing the incidence of and mortality from this disease. However, their effectiveness is limited by their low sensitivity and reproducibility. Moreover, the rare incidence of HPV-negative cancers supports the need for HPV testing for cervical cancer screening. Used either alone or in combination with cytology, HPV testing is a particularly efficient screening tool [4,5]. However, despite exhibiting better sensitivity and reproducibility than cytology [6], HPV tests have poor positive predictive value of HPV detection because only a few women with HPV will progress to a cervical lesion [7].
Currently, high-grade SIL (HSIL) is the widely accepted cut-off for treatment, because a large number of low-grade SIL (LSIL) and lower lesions spontaneously regress [8]. Following this guideline, using diagnostic tests of HPV infection for cervical cancer screening may produce too many false-positive results and lead to needless invasive procedures, prolonged follow-up periods, and psychosocial problems such as anxiety [9]. Recently, the Cobas HPV test (Roche Molecular System, Pleasanton, CA), an automated real-time polymerase chain reaction method using HPV 16 and HPV 18 genotype information, has been introduced with Food and Drug Administration approval. In the ATHENA study, although the Cobas HPV test was more sensitive than cytology in detecting high-grade squamous intraepithelial lesions or cervical cancer (HSIL+), the positive predictive value for HSIL+ was from 10.1% (high-risk HPV) to 15.5% (HPV 16, 18, or both), which meant that most women with positive HPV infection did not need invasive diagnostic procedures [10]. Therefore, there is a need to identify new, more specific biomarkers to confirm the subgroup of women with HSIL or worse (HSIL+) who require treatment from the group of women with high-risk HPV infections.
Viral load and physical state estimations are very attractive candidate biomarkers for HSIL+ based on HPV-associated carcinogenesis. Some previous cross-sectional and longitudinal studies have demonstrated an association between high viral load and risk of HSIL+ [11-13], whereas other reports did not find a strong correlation [14]. Integration into the human genome is also known to be a main event in cervical carcinogenesis [15] and is considered an important viral marker [12]. Previous studies on the viral load and integration of HPV 16 alone or in combination have been performed, yielding variable results [14-17]. Such variability may result from various factors, such as using semi-quantitative methods like Hybrid Capture 2 (HC2), heterogeneous specimen quality, different methods of calculating viral load, non-typespecific evaluation of viral load, and the presence of multiple infections as confounding factors. HC2, used in many previous studies, does not determine viral load considering the cellular input and cannot distinguish between HPV types or discriminate monotypic infections from infections with multiple HPV types [16,17]. Most studies that did focus on HPV type looked only at HPV 16 or HPV 18 [11,13]. Only a few small studies have addressed the viral load of other HPV types, reporting that type-specific viral loads and clinical significance were different, type-dependent markers for HSIL+ [12,18]. In our previously published report [19], HPV 58 was found to be the third most common HPV type related to HSIL+ and almost the same number of subjects were confirmed to have HPV 58 infection. In this study, we investigated whether viral load or integration status could be useful diagnostic biomarkers for HSIL+ using type-specific evaluation of monotypic HPV 16, 18, and 58 infections.
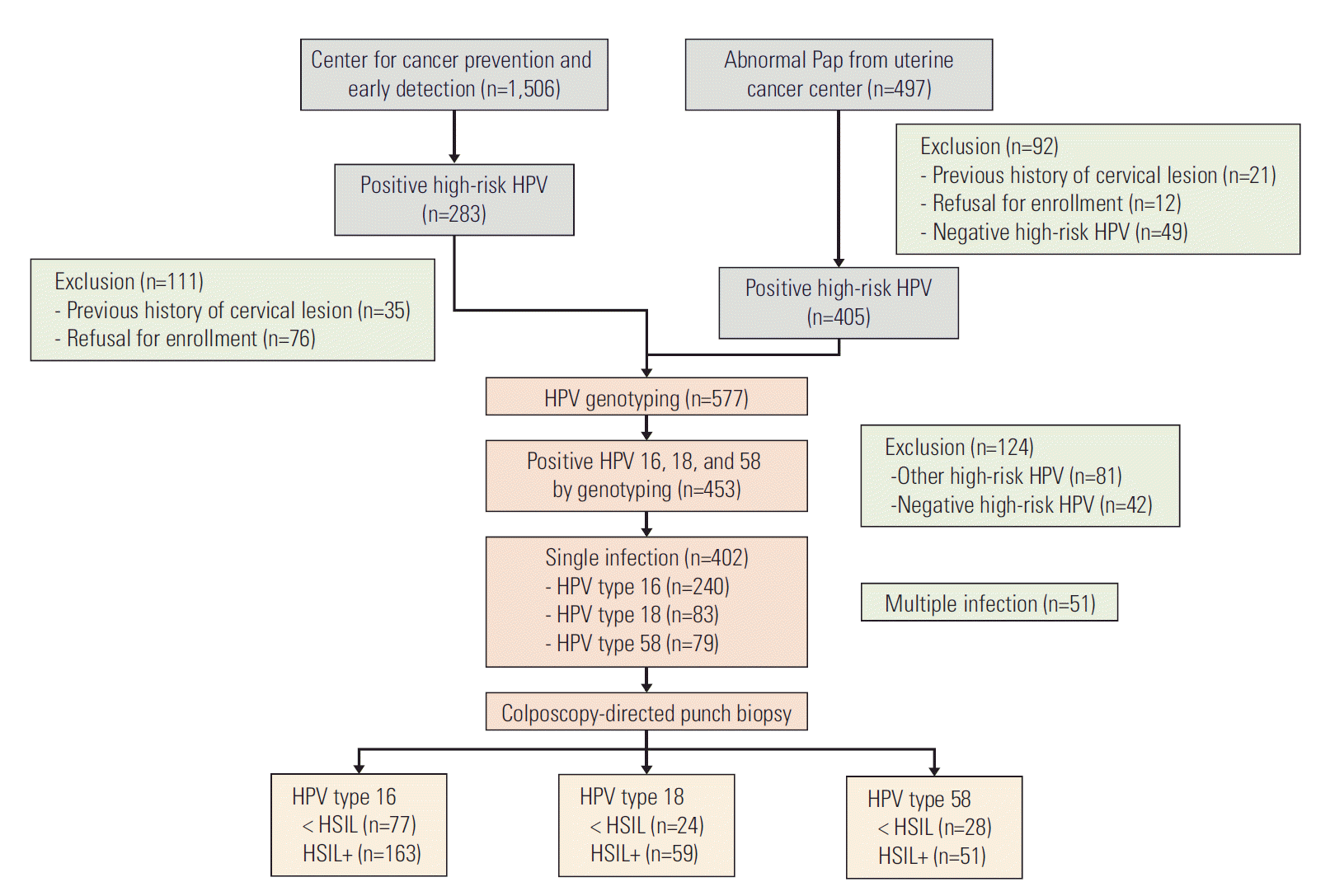
Materials and Methods
1. Study subjects
We selected 497 patients who were evaluated and treated for cervical neoplasms at the Center for Uterine Cancer. Women referred for abnormal cytologic results were considered tentative cases. Tests using the HC2 (Qiagen, Hilden, Germany) high-risk HPV kit were performed, and cervical cells were collected before colposcopy-directed biopsies were obtained. Ninety-two patients with a previous history of hysterectomy or conization, with negative results on a high-risk HPV test using HC2, or who refused to enroll in the study were excluded. An additional 1,506 women who received cervical cytologic examinations and HC2 at the Center for Cancer Prevention and Early Detection were screened during the same period. High-risk HPV infections were found in 283 women. After exclusion of 111 women with a previous history of hysterectomy or conization or who refused to enroll, 172 women were referred to the Uterine Cancer Center. Among the 577 women with high-risk HPV infections, 453 women were confirmed to be infected with HPV 16 (n=240), 18 (n=83), or 58 (n=79) by polymerase chain reaction (PCR)–based genotyping (Fig. 1). For clarity, individuals with multiple high-risk HPV infections were excluded from the analysis. Pathologic confirmation was performed by colposcopy-directed punch biopsy. We defined cases as HSIL+ because these are the targets of cervical cancer screening. Individuals without pathologic abnormalities, koilocytotic atypia, and LSIL were regarded as controls. All participants signed an informed consent form.
2. HPV genotyping
Cervical cells were centrifuged immediately after collection at 3,000 ×g and the resulting pellet was diluted in 1 mL of 1× phosphate-buffered saline and stored at –70°C until DNA extraction. DNA was isolated using the AccuPrep genomic DNA extraction kit according to the manufacturer’s instructions (Bioneer, Daejeon, Korea) within 1 week of sample collection and stored at –70°C until use. Analysis of HPV genotypes was carried out using PCR-based dot blot hybridization with HPV type-specific oligonucleotide probes [20]. HPV DNA was amplified using MY09/11 and GP5+/6+ L1 consensus primers with AmpliTaq Gold polymerase as previously described [21]. Thermocycling conditions were as follows: initial denaturation at 95°C for 9 minutes, 40 cycles at 95°C for 60 seconds, annealing at 55°C for 60 seconds, and extension at 72°C for 60 seconds, and a final 5-minute extension at 72°C. The MY09/11 and GP5+/6+ systems amplify approximately 450- and 150-base pair segments in the L1 region, respectively. During each PCR run, samples were tested together with one negative control (water) and one positive control (cells harboring HPV 16). β-Globin gene was amplified from each sample for the confirmation of presence and amount of DNA. PCR products were analyzed using gel electrophoresis and then dot blot hybridization was conducted as described previously [20]. Briefly, the PCR products were denatured in 0.4 mol/L sodium hydroxide and 10 mmol/L EDTA and applied to a nylon membrane that was premoistened with 6× saline sodium citrate (SSC) using a 48-well dot blot apparatus (Life Technologies, Grand Island, NY). DNA was then UV cross-linked to the membrane and dried. The nylon membrane was placed in prehybridization buffer(6× SSC, 0.5% sodium dodecyl sulfate [SDS], 1 mmol/L EDTA, and 100 μg/mL denatured salmon sperm DNA) for 2 hours at 68°C, followed by overnight incubation at 42°C in hybridization buffer mixed with biotin-labeled type-specific oligonucleotide probes for 16 high-risk HPV types (16, 18, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, and 69). After washing with 2× SSC and 0.5% SDS, bound probes were detected with an enhanced chemiluminescence reagent (Amersham Pharmacia Biotech, Buckinghamshire, UK). Specimens that were positive for HPV DNA but did not match any of the 16 probes were labeled high-risk HPV-negative.
3. Viral load determination using real-time PCR
Quantification of β-actin and type-specific HPV E6 and E2 genes was performed with an ABI Prism 7900HT System and TaqMan Universal PCR Master Mix (PE Applied Biosystems, Perkin-Elmer, Foster City, CA). Sequence information of each primer used is available in S1 Table. For determination of integrated viral load, the primers and probes were designed for targeting the E2 hinge regions, which are known to be deleted most frequently during viral integration [22]. β-Actin DNA loads were used for normalization. Fifty nanograms of DNA from each sample was used in each reaction and standard curves were produced by amplification of a dilution series of 109-10 copies of a clone of type-specific HPV or β-actin DNA in the pGEM-T vector. Control reactions without template were included in each test and all experiments were performed in triplicate. The mean value was used if the three values were similar, whereas the mean of two values was used if the third was an outlier. If the test was negative, direct sequencing of the PCR product with the L1 consensus primer was performed to confirm the HPV genotype. If the sample was confirmed as HPV 16, 18, or 58, quantification was repeated. We used the following formula to convert plasmid DNA into copy number: (number of base pairs)× (average weight of double-stranded DNA)/Avogadro’s number=g/plasmid molecule, (g/μL)/(g/plasmid molecule)=plasmid molecules/μL or copies/μL. The viral load was normalized to cellular DNA input as indicated below: HPV DNA load (HPV copies/cell)=number of HPV copies/(number of β-actin copies/2) [23,24]. A normalized HPV load over the 80th percentile of controls was considered a high viral load.
4. Determination of the physical state of HPV
The physical state of HPV was determined using normalized viral loads of type-specific HPV E2 and E6. The integrated viral load was determined by subtracting normalized E2 load from normalized E6 load. Then, the physical state was calculated by the ratio of integrated load (E6–E2)/total load (E6), which varied from 0 to 1. The definition of integrated physical state was an (E6–E2)/E6 ratio greater than 0.8. A ratio of 0.8 or less was considered episomal.
5. Statistical analyses
Continuous variables were assessed for normality distribution (Kolmogorov–Smirnov test) and are expressed as median with interquartile ranges. Analyses using the KruskalWallis or the Mann-Whitney U tests were performed to determine statistical significance as appropriate. Categorical variables were evaluated via chi-square analysis and logistic regression analysis to obtain odds ratios (ORs). Receiver operating characteristic (ROC) analyses and determination of the area under the curve (AUC) were performed to evaluate whether a parameter could differentiate between HSIL+ and controls. Three replicates were used to monitor the performance of each experiment. p < 0.05 was considered statistically significant. IBM SPSS Statistics ver. 21.0 (IBM Corp., Armonk, NY) was used for all statistical analyses.
Results
1. Difference in viral loads according to HPV type and SIL pathologic severity
The distribution of HPV 16, 18, and 58 viral loads in a cell with the same pathologic grade of cervical lesion is shown in Fig. 2. We observed that total viral loads were different from type to type in lesions with the same pathologic grade (p < 0.001). The viral load of HPV 58 was the highest, whereas the viral load of HPV 18 was the lowest (Table 1). The normalized total viral loads of HPV 16, 18, and 58 (copy/cell) increased with the pathologic severity of SIL (p < 0.001) (Table 1).
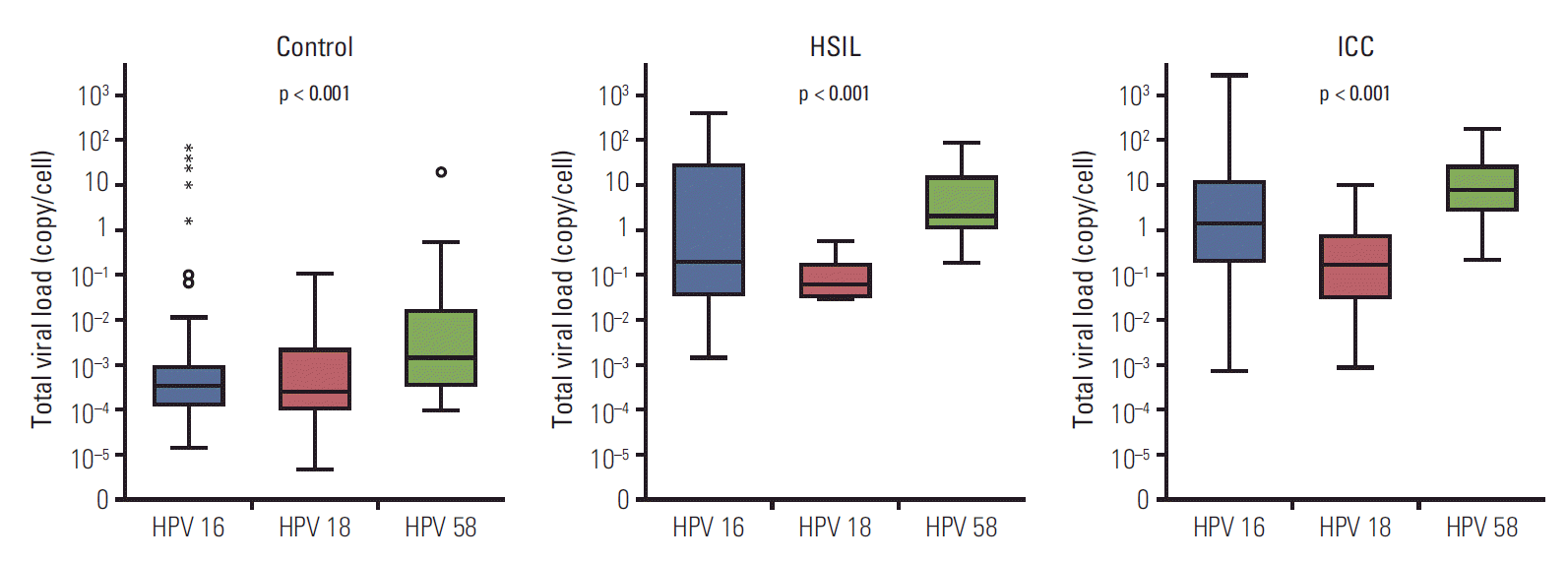
Fig. 2.
Type-specific normalized total viral load of human papillomavirus (HPV) 16, 18, and 58 in patients with cervical lesions at the same pathologic grade. Box plots show median and interquartile range. Controls include patients without pathologic abnormalities or with low-grade squamous intraepithelial lesions. p-values were evaluated using Kruskal-Wallis tests. HSIL, high-grade squamous intraepithelial lesions; ICC, invasive cervical cancer.
![]()
Table 1.
Type-specific total viral load and pathologic grade of cervical lesion
| HPV type |
HPV 16 |
HPV 18 |
HPV 58 |
p-value | |||
|---|---|---|---|---|---|---|---|
| No. | Viral load (copy/cell) | No. | Viral load (copy/cell) | No. | Viral load (copy/cell) | ||
| Control | 77 | 0.000 (0.000-0.001) | 24 | 0.000 (0.000-0.002) | 28 | 0.002 (0.000-0.020) | < 0.001 |
| HSIL | 36 | 0.208 (0.034-31.177) | 6 | 0.062 (0.032-0.272) | 15 | 2.087 (1.006-25.000) | < 0.001 |
| ICC | 127 | 1.505 (0.202-11.724) | 53 | 0.175 (0.032-0.769) | 36 | 7.664 (2.716-26.433) | < 0.001 |
| p-valuea) | < 0.001 | < 0.001 | < 0.001 | ||||
![]()
2. Type-specific total and integrated viral load and physical state of HSIL+
The median total viral loads of HPV 16, 18, and 58 were significantly higher in the HSIL+ group than in controls (p < 0.001 for all) (Table 2). In addition, the median integrated viral loads of HPV 16, 18, and 58 were significantly higher in HSIL+ cases than in controls (p < 0.001 for HPV 16 and 18; p=0.007 for HPV 58). Logistic regression analysis revealed that high total viral load was an independent risk factor for all HPV types. The ORs of high viral load of HPV 16, 18, and 58 were 14.065 (95% CI, 5.126 to 38.634), 39.472 (95% CI, 2.814 to 554.821), and 7.103 (95% CI, 2.523 to 20.005), respectively (Table 3). However, high integrated viral load was an independent risk factor only for HPV 16 (OR, 8.242; 95 % CI, 2.633 to 25.825), and integration into the host genome was an independent risk factor only for HPV 18 (OR, 9.443; 95 % CI, 1.454 to 61.605).
Table 2.
Differences in type-specific viral loads in the HSIL+ group
![]()
Table 3.
Logistic regression analysis of type-specific viral loads and physical state for detection of HSIL+
![]()
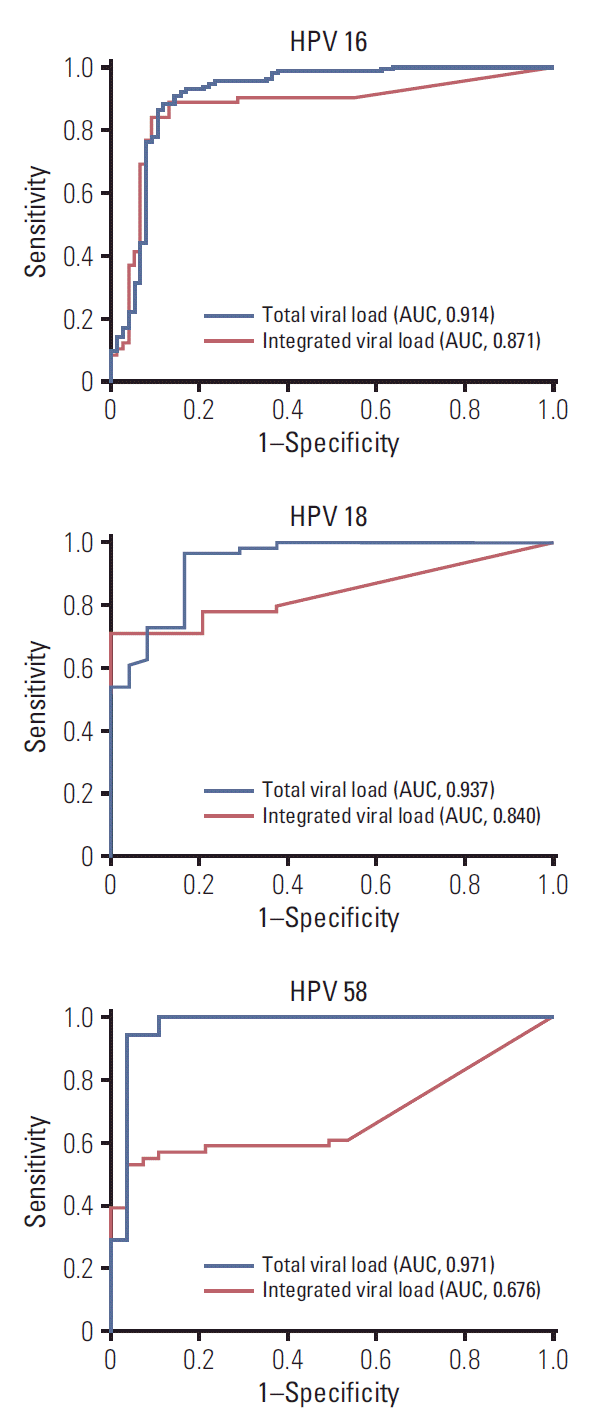
3. ROC analysis of viral load as a diagnostic biomarker of HSIL+
We next evaluated whether type-specific viral load could differentiate between the control and HSIL+ groups. ROC curves for controls versus HSIL+ are shown in Fig. 3. Type-specific viral load was associated with HSIL+ (AUC, 0.914, 0.937, and 0.971 for HPV 16, 18, and 58, respectively), indicating that it has excellent diagnostic value in differentiating HSIL+ cases from controls. For integrated viral load, the AUCs of HPV 16, 18, and 58 were 0.871 (95% CI, 0.818 to 0.924), 0.840 (95% CI, 0.757 to 0.922), and 0.676 (95% CI, 0.560 to 0.793), respectively, and thus inferior to total viral load as a diagnostic biomarker of HSIL+.
Discussion
We have shown here that type-specific normalized total viral load is a powerful diagnostic biomarker of HSIL+. This observation may help overcome the limitations of using HPV testing for cervical cancer screening.
The present study has several advantages over previous studies. First, we normalized viral load to cellular input DNA, which is a very objective and reproducible method. Many studies reported that high HPV viral load is associated with persistent infection and may be useful in predicting HSIL+, but the proposed cut-off values of viral load differed greatly [12-14,24,25]. These different cut-off values result from various factors including differences in experimental methods and different methods of estimating viral load. Because the ratio between the number of HPV DNA copies and the number of cells in the tested specimen seems the most credible and reproducible means of measuring viral load, we used the number of copies of HPV DNA per cell as an expression of viral load. We suggest that normalizing viral load to cellular DNA input can produce reliable and reproducible findings for clinical applications. Specimen homogeneity, including absolute amount of input DNA, may be an important issue in this kind of study, because the number of cells in the specimen may affect viral load quantification [26]. The range of viral load overlaps with the pathologic group if the degree of specimen heterogeneity is significant. The viral loads determined in our results were relatively well quantified by normalization to cellular input DNA. Second, we analyzed the diagnostic roles of viral load and physical state stratified by type. The few, small studies that have been performed on viral loads of other HPV types reported that viral loads were type-dependent markers for HSIL+ [12,18]. However, most studies on viral load and HPV integration focused only on type 16 or pooled data irrespective of type [11,27], a few looked at type 18 [13,28,29], and very few examined other types [18,30]. This is because sample size is a major obstacle to accurate evaluation of the role of type-specific viral load except in the case of HPV 16. The number of study subjects infected only with HPV 18 or 58 in the current study is the largest to our knowledge, and thus our findings are more reliable in assessing the effect of viral load and its diagnostic value in cervical cancer screening. HPV 58 is a high-risk type with low prevalence in Eastern countries but considerably more prevalent in Asian countries [19,30]. There have been very few studies on the correlation between HPV 58–specific viral load and integration and disease severity [18]. Although previous studies related to HPV 58 produced unclear results, our study supports the usefulness of viral load as a diagnostic marker of HSIL+, but not integration or physical status. Compared with previous studies, our study featured a larger sample size, fewer confounders (as subjects had only a single infection), and colposcopy-directed biopsy confirmation. From our findings, we can conclude that the mechanism of carcinogenesis differs depending on HPV type and the effect of viral load and physical state may differ from type to type. Thus, type-specific analysis is desirable for accurate evaluation. Third, viral load and integration, the most important viral markers of HSIL+, were considered at the same time. Although not only total viral load but also integrated viral load seem to be valuable for the diagnosis of HSIL+ in subjects with HPV 16, only total viral load, not integrated viral load, was useful in cases of HPV 18 or 58 in our results.
A few recent studies observed higher viral loads of HPV 18 in subjects with higher pathologic grade cancers [28,29], whereas many other studies failed to observe a significant effect of HPV 18 viral load on disease severity [13]. In our results, the viral load of HPV 18 was significantly lower than that of HPV 16 and 58, and was distributed more narrowly, than that of HPV 16 or 58. These results are consistent with a recently published report using the Cobas HPV test with normalization [29]. However, the authors could not find a linear relationship of HPV 18 viral load, but only increased viral loads in invasive cervical cancer (ICC) compared to HSIL [29]. This discrepancy may result from the small sample size in both studies. In the report by Wu et al. [29], the number of HPV 18–positive HSIL and ICC samples was only 10 and 27, respectively. Similarly, in our study, the number of HPV 18–positive HSIL and ICC samples was only 6 and 53, respectively. Although we found a statistical difference in viral load according to pathologic severity, it seems insufficient for a final decision on linear trends considering these controversial results. Assuming that specific adjuvant factors or carcinogenic mechanisms could result in a powerful oncogenic potential despite a low viral load, early and high-frequency integration may have an important role in HPV 18–related cervical malignancies. We found that integration state was an independent risk factor in cases of HPV 18.
Another important finding of this study is that the frequency of integration of HPV 58 in patients with HSIL+ was very low compared to that of HPV 16 or 18. Previously, integration was considered a critical event in carcinogenesis of cervical cancer as it results in loss of the episomal type. It has been reported that intact E2 expression derived from episomal HPV DNA has an inhibitory effect on viral oncogene expression [15], and integrated HPV survive during carcinogenesis because of a relative priority in cell proliferation and avoidance of the host immune response [31]. In contrast, some studies demonstrated the presence of integrated HPV 16 only in 28% to 67% of subjects [3], and the frequency of integration was markedly different in cases with different HPV types [32]. For HPV 58, the integration frequency in cervical cancer was only 8.3% in our results, which is in agreement with data previously reported by Ho et al. [18], who found the integrated from of HPV 58 in only 12.5% of cervical cancers. Our findings are also similar to those of a study that reported that the integration frequency of HPV 18 is the highest of all the types [18]. The high prevalence of the episomal form in HPV 58–related cervical cancer supports the conclusion that E2 disruption is not mandatory for enhanced oncogene expression, and the incidence of integration probably is different from type to type. There is a need to investigate alternative mechanisms of going beyond a limit during the carcinogenesis of HPV 58. There have been possible explanations for the episomal form of HPV 58, including the significant proportion of HPV 16 in cervical cancers. These include epigenetic controls such as methylation of viral and host DNA and mutations in the long control region, which may lead to the acquisition of invasiveness [27]. Differences in the frequency of integration of HPV types suggest that integration is not a precondition for cervical carcinogenesis, nor is it adequate as a diagnostic tool irrespective of HPV type.
The present study has some limitations. First, the sample size of subjects infected with HPV 18 or 58 was small. Especially for HPV 18, the HSIL group had only six samples, and the differences in viral load between HSIL and ICC was not large compared to that of HPV 16 (2.5 times and 7.2 times, respectively). Considering the controversial results relative to linear relationship of viral load according to disease severity [29], a larger number of subjects will be needed to resolve this issue. Second, we performed a cross-sectional study. Although our results show that type-specific viral load is correlated to the pathologic grade of cervical neoplasia, these results cannot be used to predict the risk of disease progression. Clinically, predicting the risk of lesion progression is quite important, considering that a large proportion of cervical intraepithelial neoplasia cases spontaneously regress. Longitudinal studies that investigate both the load and physical state of HPV are needed to evaluate their usefulness in predicting the risk of cervical cancer in the future. Third, it is both a strong and a weak point that our subjects included only those with a single type of infection, because significant proportions of HSIL+ individuals have multiple infections. Which factor, specific HPV type or viral load, is more reliable in predicting HSIL+ needs to be examined in the future. Lastly, we cannot draw any conclusions regarding the diagnostic value of HPV types other than 16, 18, and 58, as we did not look into other types. Large-scale trials are necessary, because the incidence of other types is low enough to present a statistical challenge. We are now performing a multicenter trial with the help of the Korean Gynecologic Oncology Group to reach this goal. The technical development of molecular testing of HPV using real-time PCR have resulted in an increasingly advantageous method. Similarly to the Cobas HPV test, recent HPV tests using real-time PCR can provide multiple results in one test run with multiple fluorescent dyes. Once the clinical relevance of viral load is confirmed, commercially useful HPV tests using viral load information will be developed.
In conclusion, type-specific total viral load can be a useful diagnostic biomarker of HSIL+, at least in HPV 16–, 18–, and 58–infected women. Collectively, our findings can help increase the diagnostic accuracy of HPV tests, leading to a paradigm shift in the management of equivocal cytologic abnormalities in cervical cancer screening.
 XML Download
XML Download