

 PDF
PDF Citation
Citation Print
Print


Introduction
Imatinib is the first tyrosine kinase inhibitor (TKI) approved for the treatment of chronic myeloid leukemia (CML), Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL), and gastrointestinal stromal tumors (GIST) [1]. Its mechanism of action involves preventing the phosphorylation of the cell cycle-regulating substrate by occupying the adenosine triphosphate-binding site of several tyrosine kinase molecules including Bcr-Abl, c-KIT, and platelet-derived growth factor receptors (PDGFRα and PDGFRβ) [2,3].
Various adverse effects of imatinib have been reported. The common adverse effects include nausea, fluid retention, muscle cramps, diarrhea, and vomiting, which are generally mild to moderate in severity and manageable [2]. Elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) is another important adverse effect although it occurs relatively less frequently [4]. Clinical studies have reported that the incidence of a grade Ⅱ or higher abnormality in serum aminotransferases was about 5% and the incidence of grade Ⅲ-Ⅳ elevation was 1.0% to 5.1% [2,5,6].
Hepatotoxicity of imatinib could aggravate the patient’s clinical condition and alter the patient’s treatment plan. Permanent imatinib discontinuation due to hepatotoxicity is necessary in 0.5% of patients [6]. Fatal cases associated with severe hepatotoxicity have been reported including acute hepatitis, severe hyperbilirubinemia, and focal necrosis, which led to liver failure and death [7,8]. Therefore, the identification of risk factors for imatinib-induced hepatotoxicity could reduce the occurrence of hepatotoxicity, thereby preventing progression to chronic liver disease and/or acute liver failure. However, the factors for imatinib-induced hepatotoxicity have rarely been investigated.
In general, it was reported that 62% of drug-induced hepatotoxicity cases occurred within one month from medication commencement [9]. In the case of imatinib, a review article described that the elevation of transaminase levels were generally observed during the first 2-3 months after imtinib initiation [10]. In addition, another study showed that the latency to onset of hepatic injury by imatinib was 12 to 77 days [11]. Based on such information from previous literature regarding the onset of imatinib-induced hepatotoxicity, the aim of this study was to investigate factors affecting the incidence of hepatotoxicity within 90 days after starting imatinib treatment and time to onset of imatinib-induced hepatotoxicity.
Go to : 
Materials and Methods
1. Patients
This retrospective study was performed using medical records from October 2012 to September 2017 at Seoul National University Hospital, Korea. Eligible patients were older than 18 years and received imatinib for treatment of Philadelphia chromosome-positive ALL, CML, GIST, and other malignancies. Patients were excluded if they already had elevated AST/ALT or were concurrently receiving hepatotoxic anti-cancer drugs such as cyclophosphamide, cytarabine, etoposide, methotrexate, and vincristine.
The following demographic and clinical data were collected: sex, age, body weight, body surface area (BSA), underlying disease, imatinib daily dose, and concomitant medications. Concomitant medications included cytochrome P450 (CYP) 3A4 inhibitors, CYP3A4 inducers, H2-antagonists, and proton pump inhibitors (PPIs). CYP3A4 inhibitors included aprepitant, ciprofloxacin, fluconazole, nicardipine, nifedipine, posaconazole, and tamoxifen. CYP3A4 inducers included clarithromycin, dexamethasone, and rifampicin (rifampin). H2-antagonists included famotidine and ranitidine. PPIs included (es)omeprazole, lansoprazole, and pantoprazole.
2. Administration and laboratory assessment
Patients received imatinib (dose range, 100 to 800 mg/day) orally. Serum AST and ALT levels were obtained before initiation of therapy and every month thereafter. The hepatotoxicity grade was determined using Common Terminology Criteria for Adverse Events (CTCAE), ver. 4.0. The CTCAE defines grade Ⅰ, grade Ⅱ, grade Ⅲ, and grade Ⅳ toxicity levels of AST and ALT as 1 to 3 times, 3 to 5 times, 5 to 20 times, and more than 20 times the upper limit of normal, respectively. In this study, hepatotoxicity was defined as grade I or higher.
3. Statistical analysis
The chi-square or Fisher exact test was used to compare categorical variables between patients with and without hepatotoxicity. Multivariate logistic regression analysis was used to identify independent risk factors for hepatotoxicity. Factors having a p-value less than 0.2 from the univariate analysis along with the strong confounders of sex, age, and BSA were included in the multivariate analysis. Odds ratio (OR) and adjusted OR were estimated from univariate and multivariate analyses, respectively. Attributable risk (%) was calculated as (1–1/OR)×100. The time to reach hepatotoxicity was analyzed using Kaplan-Meier survival curves and the log-rank test. The Cox proportional-hazards model was used for the multivariate analysis. Factors having a p-value less than 0.2 from the univariate analysis along with the strong confounders of sex, age, and BSA were included in the multivariate analysis. Hazard ratio (HR) and adjusted HR were calculated from the univariate and multivariate analyses, respectively. p-values less than 0.05 were considered statistically significant.
To test the model’s goodness of fit, we performed a Hosmer-Lemeshow test. Discrimination of the model was further analyzed by the area under the receiver operating characteristic curve (AUROC) to assess the ability of the risk factor to predict hepatotoxicity related to imatinib administration. All statistical analyses were performed with the SPSS ver. 20.0 for Windows (IBM Corp., Armonk, NY).
4. Ethical statement
This retrospective study was approved by the Clinical Research Ethics Committee of the Seoul National University Hospitals, Korea (IRB No. 1711-061-899). The requirement for informed consent from patients was waived.
Go to :
Results
A total of 301 patients treated from October 2012 to September 2017 were eligible for participation in the study. We excluded patients who did not have AST/ALT level results before administration of imatinib (n=4) and who had elevated AST/ALT levels on the first day of imatinib administration (n=98). Patients who received concomitant anti-cancer drugs were also excluded (n=22). Consequently, data from 177 patients with imatinib administration were analyzed.
As shown in Table 1, 101 patients (57.1%) were older than 60 years (age range, 21 to 90 years). Approximately 51% of the study patients were male. Drugs concurrently administered with imatinib were CYP3A4 inducers (n=1), CYP3A4 inhibitors (n=4), H2-antagonists (n=16), PPIs (n=18), and any of the two anti-acid secreting agents (H2-antagonist/PPI, n=32). The proportion (number) of patients with hepatotoxicity within 90 days after initiation of imatinib administration was 33.9% (n=60). Among 60 patients with hepatotoxicity, the number of patients with grade II and grade III hepatotoxicity was three each, while the other 54 patients experienced grade I hepatotoxicity. In the univariate analysis, co-administration of PPIs, body weight under 55 kg, and presence of liver disease or hepatitis B virus (HBV) were significant factors for hepatotoxicity within 90 days.
Table 1.
Hepatotoxicity within 90 days related to imatinib administration
| Characteristic | No. (%) (n=177) |
Hepatotoxicity |
p-value | |
|---|---|---|---|---|
| Presence (n=60) | Absence (n=117) | |||
| Age (yr) | ||||
| < 60 | 76 (42.9) | 24 (40.0) | 52 (44.4) | 0.572 |
| ≥ 60 | 101 (57.1) | 36 (60.0) | 65 (55.6) | |
| Sex | ||||
| Male | 90 (50.8) | 31 (51.7) | 59 (50.4) | 0.876 |
| Female | 87 (49.2) | 29 (48.3) | 58 (49.6) | |
| Body weight (kg)a) | ||||
| < 55 | 72 (43.9) | 33 (56.9) | 39 (36.8) | 0.013 |
| ≥ 55 | 92 (56.1) | 25 (43.1) | 67 (63.2) | |
| BSA (m2)b) | ||||
| < 1.6 | 83 (50.9) | 35 (60.3) | 48 (45.7) | 0.074 |
| ≥ 1.6 | 80 (49.1) | 23 (39.7) | 57 (54.3) | |
| CVD or DM | ||||
| Yes | 69 (39.0) | 21 (35.0) | 48 (41.0) | 0.437 |
| No | 108 (61.0) | 39 (65.0) | 69 (59.0) | |
| Liver metastasis | ||||
| Yes | 20 (11.3) | 5 (8.3) | 15 (12.8) | 0.372 |
| No | 157 (88.7) | 55 (91.7) | 102 (87.2) | |
| Presence of liver disease or HBV | ||||
| Yes | 11 (6.2) | 7 (11.7) | 4 (3.4) | 0.031 |
| No | 166 (93.8) | 53 (88.3) | 113 (96.6) | |
| Daily dose (mg) | ||||
| ≤ 400 | 166 (93.8) | 55 (91.7) | 111 (94.9) | 0.403 |
| > 400 | 11 (6.2) | 5 (8.3) | 6 (5.1) | |
| CYP3A4 inducer | ||||
| Yes | 1 (0.6) | 0 | 1 (0.9) | 0.473 |
| No | 176 (99.4) | 60 (100) | 116 (99.1) | |
| CYP3A4 inhibitor | ||||
| Yes | 4 (2.3) | 1 (1.7) | 3 (2.6) | 0.704 |
| No | 173 (97.7) | 59 (98.3) | 114 (97.4) | |
| PPI | ||||
| Yes | 18 (10.2) | 11 (18.3) | 7 (6.0) | 0.010 |
| No | 159 (89.8) | 49 (81.7) | 110 (94.0) | |
| H2-antagonist | ||||
| Yes | 16 (9.0) | 4 (6.7) | 12 (10.3) | 0.430 |
| No | 161 (91.0) | 56 (93.3) | 105 (89.7) | |
| H2-antagonist/PPI | ||||
| Yes | 32 (18.1) | 13 (21.7) | 19 (16.2) | 0.374 |
| No | 145 (81.9) | 47 (78.3) | 98 (83.8) | |
![]()
Univariate analysis showed that patients who took PPIs with imatinib had a 3.5-fold increased incidence of hepatotoxicity than those not using PPIs (Table 2). Also, patients with body weight < 55 kg had a 2.3-fold increased incidence of hepatotoxicity than those with body weight ≥ 55 kg. Presence of liver disease or HBV increased hepatotoxicity by 3.7 times. Because BSA and body weight are co-related, two models were conducted for multivariate analysis. Model I included the presence of liver disease or HBV and PPIs in addition to sex, age, and BSA; model II included body weight instead of BSA. Based on both models, PPIs and the presence of liver disease or HBV increased the incidence of hepatotoxicity approximately 3.8-fold and 8.5-fold, respectively, after controlling for variables with p-values less than 0.2 from the univariate analysis. Patients with body weight under 55 kg had a 2.2-fold increased incidence of hepatotoxicity. The Hosmer-Lemeshow test for multivariate models revealed good fit (χ2=0.395, p=0.821 for model I and χ2=4.201, p=0.122 for model II). As shown in Fig. 1, the AUROC was 0.650 (95% confidence interval [CI], 0.559 to 0.740) for model I and 0.657 (95% CI, 0.566 to 0.748) for model II.
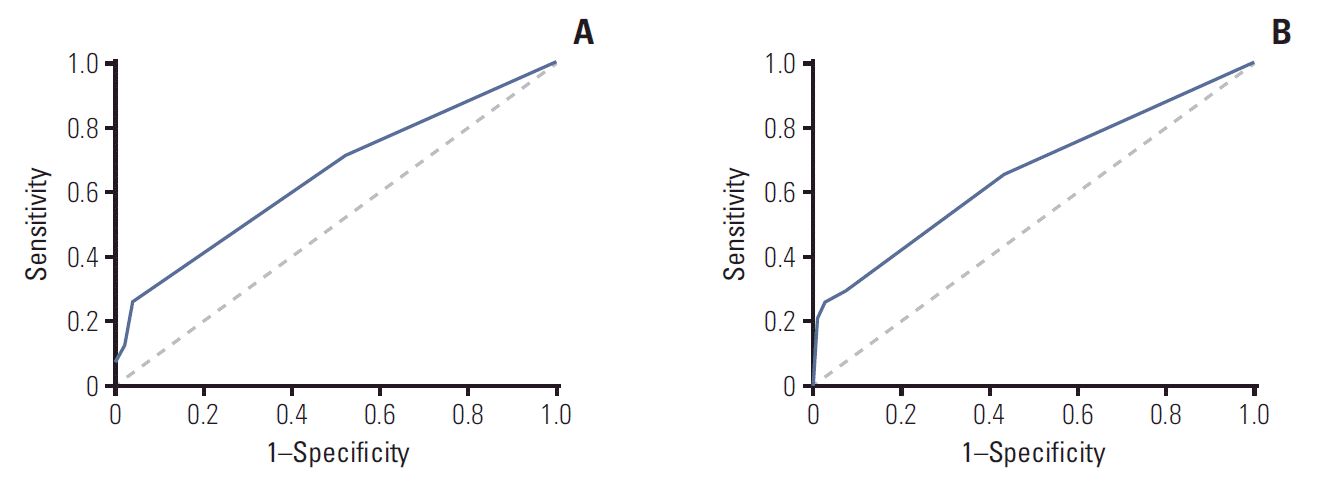 | Fig. 1.Area under the receiver operating characteristic curve for factors affecting imatinib-induced hepatotoxicity. (A) Model I included for analysis the body surface area, presence of liver disease or hepatitis B virus infection, and proton pump inhibitors. (B) Model II included for analysis the body weight, presence of liver disease or hepatitis B virus infection, and proton pump inhibitors.
|
Table 2.
Univariate and multivariate analyses to identify predictors for hepatotoxicity related to imatinib administration within 90 days
| Characteristic | Unadjusted OR (95% CI) |
Model Ⅰ |
Model Ⅱ |
||
|---|---|---|---|---|---|
| Adjusted OR (95% CI) | Attributable risk (%) | Adjusted OR (95% CI) | Attributable risk (%) | ||
| Female sex | 0.952 (0.511-1.774) | - | - | - | - |
| Age ≥ 60 yr | 1.200 (0.638-2.258) | - | - | - | - |
| BSA < 1.6 m2 | 1.808 (0.943-3.460) | 1.981 (0.992-3.953) | 49.52 | - | - |
| BW < 55 kg | 2.268 (1.181-4.348)* | - | - | 2.247 (1.134-4.464)* | 55.50 |
| Presence of liver disease or HBV | 3.731 (1.047-13.300)* | 8.744 (1.685-45.383)* | 88.56 | 8.292 (1.586-43.363)* | 87.94 |
| PPI | 3.528 (1.290-9.644)* | 3.882 (1.318-11.431)* | 74.24 | 3.806 (1.279-11.326)* | 73.73 |
For model Ⅰ construction, sex, age, BSA, presence of liver disease or HBV, and PPI were included for analysis. For model Ⅱ construction, sex, age, BW, presence of liver disease or HBV, and PPI were included for analysis. OR, odds ratio; CI, confidence interval; BSA, body surface area; BW, body weight; HBV, hepatitis B virus; PPI, proton pump inhibitor.
![]()
Among the 177 patients, 100 patients who had hepatotoxicity during the study period were evaluated for time to reach hepatotoxicity. The median time to hepatotoxicity was 70 days. Presence of liver disease or HBV and daily imatinib dose greater than 400 mg were significant factors for time to hepatotoxicity (Table 3). Multivariate analysis showed that patients taking PPIs and patients with liver disease or HBV had an approximately 2.1- and 5.2-fold increased hazard of hepatotoxicity compared to those not taking PPIs and without liver disease or HBV, respectively (Table 4). Patients who received an imatinib dose > 400 mg had a 2.3-fold increased hazard of hepatotoxicity compared to those with an imatinib dose ≤ 400 mg.
Table 3.
Time to hepatotoxicity related to imatinib administration
| Characteristic | No. (%) (n=100) | Time to hepatotoxicity Median (95% CI, day) | p-value |
|---|---|---|---|
| Age (yr) | |||
| < 60 | 40 (40.0) | 56 (34.307-77.693) | 0.760 |
| ≥ 60 | 60 (60.0) | 77 (36.537-117.463) | |
| Sex | |||
| Male | 57 (57.0) | 77 (42.828-111.172) | 0.839 |
| Female | 43 (43.0) | 70 (26.408-113.592) | |
| BW (kg)a) | |||
| < 55 | 45 (47.9) | 49 (17.452-80.548) | 0.283 |
| ≥ 55 | 49 (52.1) | 83 (40.477-125.523) | |
| BSA (m2)b) | |||
| < 1.6 | 51 (54.3) | 53 (27.010-78.990) | 0.821 |
| ≥ 1.6 | 43 (45.7) | 77 (35.883-118.117) | |
| CVD or DM | |||
| Yes | 34 (34.0) | 70 (31.210-108.790) | 0.405 |
| No | 66 (66.0) | 68 (27.197-108.803) | |
| Liver metastasis | |||
| Yes | 11 (11.0) | 98 (13.842-182.158) | 0.864 |
| No | 89 (89.0) | 70 (40.227-99.773) | |
| Presence of liver disease or HBV | |||
| Yes | 6 (6.0) | 14 (7.999-20.001) | < 0.001 |
| No | 94 (94.0) | 77 (40.894-113.106) | |
| Daily dose (mg) | |||
| ≤ 400 | 93 (93.0) | 77 (42.365-111.635) | 0.049 |
| > 400 | 7 (7.0) | 42 (0.000-85.626) | |
| CYP3A4 inducer | |||
| Yes | 0 | N.A | N.A |
| No | 100 (100) | 70 (39.715-100.285) | |
| CYP3A4 inhibitor | |||
| Yes | 4 (4.0) | 105 (0.000-288.260) | 0.762 |
| No | 96 (96.0) | 68 (40.154-95.846) | |
| PPI | |||
| Yes | 12 (12.0) | 24 (0.000-83.409) | 0.193 |
| No | 88 (88.0) | 70 (34.899-105.101) | |
| H2-antagonist | |||
| Yes | 9 (9.0) | 88 (0.000-207.794) | 0.916 |
| No | 91 (91.0) | 70 (38.572-101.428) | |
| H2-antagonist/PPI | |||
| Yes | 19 (19.0) | 88 (25.435-150.565) | 0.677 |
| No | 81 (81.0) | 70 (41.599-98.401) |
![]()
Table 4.
Univariate and multivariate analyses to identify predictors for time to hepatotoxicity related to imatinib administration
| Characteristic | Unadjusted HR (95% CI) | Adjusted HR (95% CI) |
|---|---|---|
| Female sex | 0.959 (0.636-1.445) | - |
| Age ≥ 60 yr | 0.940 (0.628-1.406) | - |
| BSA < 1.6 m2 | 0.953 (0.628-1.447) | - |
| Daily dose > 400 mg | 2.136 (0.978-4.664) | 2.252 (1.020-4.974)* |
| Presence of liver disease or HBV | 4.937 (2.074-11.749)** | 5.195 (2.151-12.544)** |
| PPI | 1.491 (0.811-2.741) | 2.117 (1.109-4.042)* |
![]()
Go to :
Discussion
The results of this study showed that PPIs and the presence of liver disease or HBV were significant factors for hepatotoxicity within 90 days after imatinib initiation and the hazard of time to reach hepatotoxicity. PPI use increased the incidence of hepatotoxicity around 3.8-fold and the hazard of time to reach hepatotoxicity 2.1-fold. Patients with liver disease or HBV carriers had more than 8-fold higher risk of hepatotoxicity and 5.2-fold increased hazard of time to hepatotoxicity compared to patients without liver disease or HBV. Patients with body weight under 55 kg had a 2.2-fold higher risk for occurrence of hepatotoxicity. Patients who received imatinib at a dose larger than 400 mg had 2.3-fold increased hazard of time to reach hepatotoxicity compared with those who received imatinib at a dose of 400 mg or less.
The presence of liver disease or HBV was a significant factor for the incidence of hepatotoxicity within 90 days. Patients with pre-existing elevated AST/ALT values were excluded from the study; the patients included in this study who had liver disease or who were HBV carriers had normal AST/ALT levels before imatinib administration. There is limited previous research on imatinib-induced hepatotoxicity in patients with hepatic impairment. However, because imatinib is metabolized in the liver, the plasma concentration of imatinib may be increased in patients with liver disease or who are HBV carriers [3]. A clinical study showed that the mean maximum concentration (Cmax)/dose of imatinib and area under the curve (AUC)/dose of imatinib in patients with severely impaired liver function increased by approximately 63% and 45%, respectively [12]. The presence of liver disease or HBV may increase the exposure of imatinib, which can lead to hepatotoxicity.
Body weight under 55 kg and dose larger than 400 mg resulted in a significantly higher occurrence of hepatotoxicity and hazard of time to hepatotoxicity. The incidence of hepatotoxicity approximately doubled in patients who received imatinib > 400 mg compared to those who received ≤ 400 mg, although statistical significance was not obtained. The dose of imatinib is usually determined by disease diagnosed and the phase of disease, not by body weight or BSA [12]. According to the report of a phase III study comparing the effects of high-dose imatinib (800 mg daily) and conventional dose imatinib (400 mg daily) in patients with unresectable or metastatic GIST, patients in the high-dose arm experienced more severe adverse events including hepatic toxicity [13]. Another study showed that there was an inverse correlation between steady-state trough levels of imatinib and body weight [14]. Therefore, it is logical that patients who have lower body weight may experience more drug-induced toxicity.
Imatinib is known to be a substrate of ABCB1 and as both a substrate and inhibitor of ABCG2, the adenosine triphosphate-binding cassette transporters [15,16]. ABCG2 acts as a drug-efflux pump and is expressed in various normal tissues including the liver [17,18]. Use of an ABCG2 inhibitor can increase the concentration of ABCG2 substrates in tissues. PPIs, which are ABCG2 inhibitors, can increase the concentration of the imatinib in the liver, thereby leading to hepatotoxicity. In addition, PPI itself can cause hepatotoxicity, although only a case report is available for evidence [19].
Other anti-acid secreting agents, H2-antagonists, are not only substrates of ABCB1 but also inhibitors. In the liver and the gastrointestinal tract, ABCB1 mediates drug export for protecting the organs [20]. However, unlike PPIs, concomitant administration of H2-antagonists was not a significant factor for hepatotoxicity in our study, possibly due to other roles of H2-antagonists relevant to imatinib metabolization, including inhibiting organic cation transporters (OCTs).
Imatinib is transported into cells via the OCT proteins, members of the solute carrier superfamily [21,22]. Among them, OCT1 influx protein is known to play a major role in mediating the uptake of imatinib [21,22]. According to the report of a previous study using cell-lines, imatinib is transported by ABCB1 and human OCT1 (hOCT1), and hOCT1 inhibitors including verapamil, amantadine, procainamide, and prazosin decreased the uptake of imatinib [22]. The H2-antagonists, such as ranitidine and famotidine used by patients in this study, are hOCT1 inhibitors [23]. Co-administration of H2-antagonists with imatinib may initially decrease the intracellular exposure of imatinib by inhibition of OCT1, thereby reducing not only therapeutic efficacy but also adverse effects including hepatotoxicity [24,25].
Meanwhile, it has been reported that anti-secreting agent such as PPIs and H2-antagonists can reduce the absorption of many TKIs such as nilotinib, gefitinib, and erlotinib by elevating the gastric pH [26]. However, imatinib is known to be affected minimally by acid-reducing agents. The concomitant administration of omeprazole and antacid did not significantly change the AUC or Cmax of imatinib [26].
Plasma concentration of imatinib is influenced by CYP3A4 inducers or inhibitors because imatinib is predominantly metabolized by CYP3A4 [2,27]. Co-administration of ketoconazole, a selective CYP3A4 inhibitor, significantly increased mean Cmax and area under the curve at 24 hours (AUC24) of imatinib by 26% and 40%, respectively [28]. A study revealed that concomitant use of rifampicin, a strong CYP3A4 inducer, decreased imatinib Cmax and AUC24 by 54% and 68%, respectively [29]. In contrast, our study failed to demonstrate that CYP3A4 inducers/inhibitors affected incidence of hepatotoxicity and time to reach hepatotoxicity. This was probably because of the small sample size; only one patient was concomitantly treated with a CYP3A4 inducer and four with a CYP3A4 inhibitor.
The Hosmer-Lemeshow test results showed that the fit of the model for factors on increasing risk of hepatotoxicity was satisfactory. Limitations of this study relate to its retrospective single-center design. In addition, most (90%) patients had grade Ⅰ hepatotoxicity, which may not affect clinical practices. However, to our knowledge, this is the first study to investigate factors influencing imatinib-induced hepatotoxicity. Further prospective studies involving a larger number of patients and multicenter design are needed to confirm these findings and determine factors for high-grade hepatotoxicity.
In conclusion, this study showed that the presence of liver disease or HBV and the concomitant use of PPIs were associated with imatinib-induced hepatotoxicity. Considering that this is a retrospective single-center study, our findings require confirmation through a large, prospective multicenter study.
Go to :
 XML Download
XML Download