

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Liver ischemia/reperfusion (I/R) injury can occur in multiple clinical settings including liver transplantation, hypovolemic shock, and cardiac arrest (CA) [1,2]. The liver is the primary organ of detoxification and subjected to many insults that can potentially cause oxidative stress [3]. Consequently, hepatic antioxidant defense mechanisms play important roles in maintaining health [4]. The pathophysiology of liver I/R injury includes a number of mechanisms including oxidative stress, activation of inflammatory cells, apoptosis, and calcium overload [5,6]. I/R insult increases the production of reactive oxygen species (ROS) in the rat liver [7-9], resulting in initiation of oxidative stress that causes profound hepatocellular injury and ultimately leads to morbidity and mortality [2,7].
ROS are produced during mitochondrial oxidative metabolism and as a cellular response to various cytokines, xenobiotic compounds, and bacterial invasion, among other factors [10]. A complex antioxidant defense system prevents injury by attenuating oxidative stress caused by ROS [7]. Oxidative stress is caused by production of ROS to an extent that exceeds the capability of antioxidant defense mechanisms [7,11]. ROS-mediated oxidative stress plays crucial roles in organ damage and hemodynamic dysfunction in post-cardiac arrest syndrome [5]. Rat livers subjected to I/R injury generate excessive levels of ROS, and ROS-mediated oxidative stress causes damage to various types of biological molecules including DNA, RNA, proteins, lipids, catecholamine, and steroids [7,11], which are directly or indirectly associated with liver injury following I/R [7].
Superoxide dismutases (SODs) are the main endogenous antioxidant enzymes that respond to ROS [12]. SODs protect cells against oxidative damage by converting superoxide anion radicals [13]. ROS levels increase after I/R, resulting in a reduction in antioxidant enzyme activity [6]. Hepatocytes are known to be resistant to injury caused by ROS because hepatocytes contain high intracellular concentrations of glutathione (GSH), SODs, catalase (CAT), and lipid soluble antioxidants [14]. However, SOD activity in the liver is reduced after ischemic insult [4].
Hypothermia treatment has been shown to have protective effects after CA in clinical and animal model studies [15-17]. One of the mechanisms by which hypothermia following CA confers protection is thought to be reduction of oxidative damage [17-19]. Namely, acute ischemic insults at normothermia induce and increase ROS formation, lipid peroxidation, and protein oxidation, but this is attenuated by hypothermia treatment [4]. Therefore, in this study, we examined mast cell-induced inflammation, superoxide anion radical production, and SOD expression in the liver over time following 5 minutes of asphyxial cardiac arrest (ACA) in rats. Furthermore, we investigated if hypothermic therapy was able to attenuate the increase in mast cells and attenuate oxidative stress in the liver following ACA.
Go to : 
MATERIALS AND METHODS
Experimental Animals and Groups
Male Sprague-Dawley rats (10 weeks of age; body weight, 300– 330 g) were obtained from the Experimental Animal Center of Kangwon National University (Chuncheon, Korea). They were maintained under pathogen-free conditions at an appropriate temperature (about 23°C) and humidity (about 60%). The experimental protocol used in this study was approved (approval No. KW-200113-1) on the basis of ethical procedures and scientific care proposed by the Kangwon National University-Institutional Animal Care and Use Committee.
Rats (total n = 101) were randomly divided into five groups: (1) a normal group (n = 5); (2) a sham-operated group under normothermia (n = 5 for each time point) not subjected to ACA with body temperature controlled at 37°C ± 0.5°C for 4 hours after return of spontaneous circulation (RoSC; NT/sham group); (3) a group under normothermia subjected to ACA with body temperature controlled at 37°C ± 0.5°C for 4 hours after RoSC (NT/ACA group; n = 7 for each time point); (4) a sham-operated group under hypothermia not subjected to ACA with body temperature controlled at 33.0°C ± 0.5°C 4 hours after RoSC (HT/sham group; n = 5 for each time point); and (5) a group under hypothermia subjected to ACA with body temperature controlled at 33.0°C ± 0.5°C for 4 hours after RoSC (HT/ACA group; n = 7 for each time point). Rats in each group were sacrificed at 6 hours, 12 hours, 1 day, and 2 days, respectively, after RoSC.
ACA Induction and CPR
ACA and cardiopulmonary resuscitation (CPR) were performed according to published procedures [20,21] with minor modification. Briefly, rats were anesthetized with 2%–3% isoflurane and mechanically ventilated to maintain respiration using a rodent ventilator (Harvard Apparatus, Holliston, MA, USA). Body temperature, peripheral oxygen saturation (SpO2), electrocardiogram, and mean arterial pressure (MAP) were monitored and are presented in Table 1.
Table 1.
Physiological variables in the NT/sham, NT/ACA, HT/sham, and HT/ACA groups
![]()
To induce ACA, 2 mg/kg vecuronium bromide (Gensia Sicor Pharmaceuticals, Irvine, CA, USA) was administered after a 5-minute stabilization period, anesthesia was stopped, and mechanical ventilation was halted. At this point in time, MAP was below 25 mm Hg, and subsequent pulseless electric activity was used to define CA [22,23]. ACA was confirmed at 3–4 minutes after injection of vecuronium bromide and allowed to occur for 5 minutes. CPR was initiated by intravenous administration of 0.005 mg/kg of epinephrine with 1 mEq/kg of sodium bicarbonate. Mechanical ventilation with 100% oxygen and mechanical chest compression were performed at a rate of 300/min until the MAP reached 60 mm Hg and electrocardiographic activity was observed. When RoSC was not detected, half the amount of epinephrine was added and CPR was performed for 1 minute. Rats that required a third round of CPR were excluded from this study.
Hypothermia Treatment
Therapeutic hypothermia was applied after RoSC according to previously published protocols [24,25]. In brief, hypothermia was achieved by cooling the body surface with isopropyl alcohol wipes, ice packs, electrical fans, and cooling blankets. Body temperature in the hypothermia groups was maintained at 33°C ± 0.5°C for 4 hours based on monitoring with a rectal temperature sensor. Rats were rewarmed to 37°C ± 0.5°C for 1 hour with pads and warming blankets.
Liver Tissue Preparation
Liver tissues were prepared according to our published method [26]. In short, rats were deeply anesthetized by intraperitoneal administration of sodium pentobarbital (60 mg/kg) (JW Pharmaceutical, Seoul, Korea). Under anesthesia, whole bodies of rats were rinsed via the ascending aorta with saline and fixed with 4% paraformaldehyde solution. Livers were isolated, cut, embedded in paraffin, and sectioned into 6 µm-thick sections. Finally, the liver sections were mounted on gelatincoated microscopy slides.
Toluidine Blue Staining
To examine the presence of mast cells in the liver, sections were stained with toluidine blue according to a published method [27]. In brief, to make fresh stock solution, toluidine blue O (1g) was dissolved in 70% isopropanol (100ml). To make the final working solution, 5ml of the toluidine blue stock was mixed with 45ml of 1% NaCl (pH 2.3). Paraffin sections were deparaffinized, incubated in the working solution for 2–3minutes, washed in distilled water, and quickly dehydrated with 95% ethanol and 100% ethanol. After cleaning with xylene, the slides were mounted with coverslips.
To analyze changes in the number of mast cells in the liver, digital images of toluidine blue-positive cells were captured from five sections per animal using a digital camera (DP72; Olympus, Tokyo, Japan) attached to a light microscope (BX53, Olympus) connected to a PC monitor. Toluidine blue positive cells were counted in a 250 μm2 square including the central vein using image analysis software (Optimas 6.5; CyberMetrics, Scottsdale, AZ, USA). The mast cell count was obtained by averaging the total counts of three different pathologists.
Measurement of Superoxide Anion
To evaluate in situ production of superoxide anions in the liver, the oxidative fluorescent dye dihydroethidium (DHE; Sigma-Aldrich, St. Louis, MO, USA) was used. DHE is oxidized by superoxide to a fluorescent product, 2-hydroxy-ethidium, in a relatively specific reaction. Histological detection of superoxide anion radicals was performed as described previously [28]. In brief, prepared liver sections were incubated with DHE (10 μmol/L) in the dark for 30 minutes at 37°C.
To detect ethidium fluorescence, stained sections were examined under an epifluorescent microscope (Olympus) in the excitation wavelength range of 520–540 nm. DHE fluorescence intensity was analyzed from seven sections per animal. Ethidium fluorescence was quantified from images using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA). DHE fluorescence intensity was expressed as a percentage relative to that in the NT/sham group (100%).
Immunohistochemistry
We used immunohistochemistry to investigate changes in endogenous antioxidant levels (SOD1 and SOD2). In brief, according to our previously published procedure [29], prepared liver sections were reacted with solutions of sheep anti-SOD1 (diluted 1:1000; Calbiochem, La Jolla, CA, USA) or sheep antiSOD2 (diluted 1:1000; Calbiochem). These sections were then reacted with a solution containing secondary antibody (diluted 1: 250; Vector Laboratories, Burlingame, CA, USA) and developed with Vectastain ABC (Vector Laboratories). Finally, sections were visualized with 3,3’-diaminobenzidine, dehydrated, and mounted with Canada balsam.
SOD1 and SOD2 immunoreactivity in each group was quantitated as described in our previous publication [26]. In short, images of SOD1 and SOD2 immunoreactive structures were captured with a light microscope (BX53, Olympus) equipped with a digital camera connected to a PC monitor (DP72, Olympus). The density of each immunoreactive structure was evaluated as relative optical density (ROD) using NIH Image 1.59 software. ROD was expressed as a percentage compared with that in the NT/sham group.
Statistical Analysis
All statistical analyses were performed using GraphPad InStat (ver. 3.05) and data are expressed as mean ± standard error of the mean. The significance of differences between groups was assessed by one-way analysis of variance followed by the post hoc Tukey test. Differences were considered significant at P < 0.05.
Go to :
RESULTS
Mast Cells
Mast cells were rarely observed around portal areas in the livers of the NT/sham group (Figure 1A), similar to what we observed in the normal group (data not shown). In the NT/ACA group, a significant increase in the number of mast cells was observed at 6 hours after ACA compared with that in the NT/sham group (Figure 1B and K), but a significant reduction was observed at 12 hours after ACA (Figure 1C, 1K). Thereafter, the number of mast cells peaked at 1 day after ACA and decreased at 2 days after ACA (Figure 1D, E, and K).
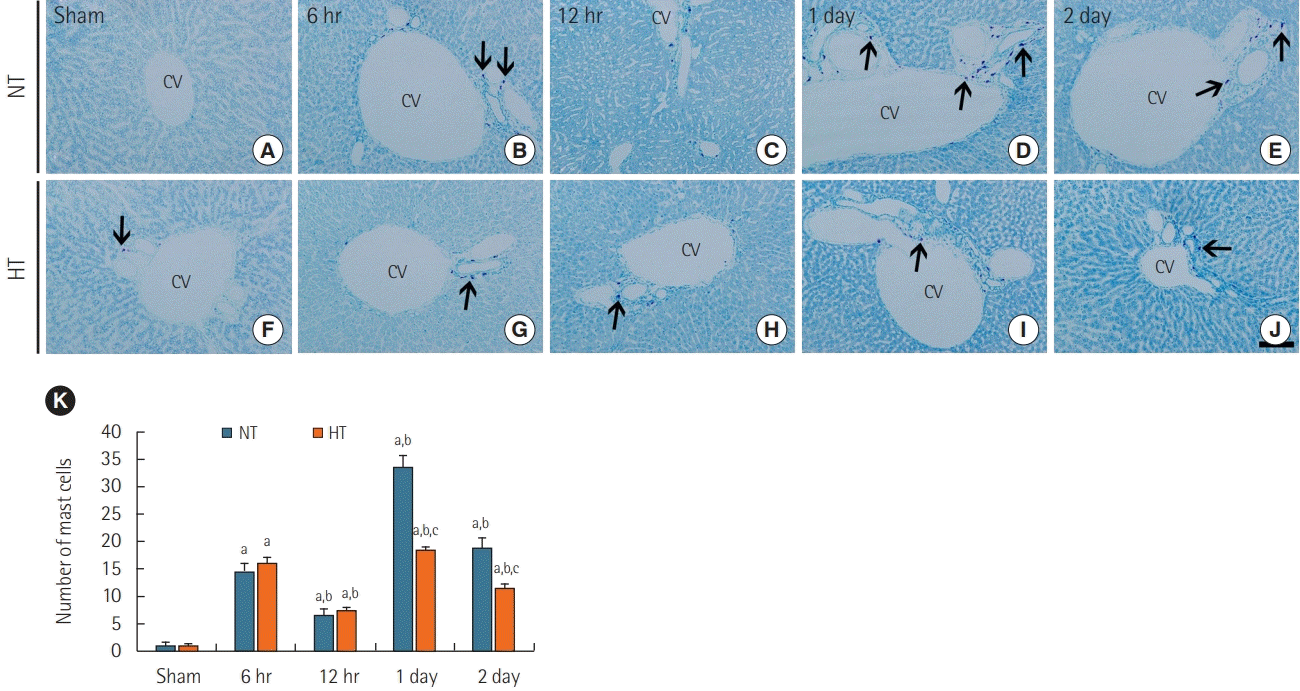 | Figure 1.Toluidine blue staining of liver sections in the normothermia (NT; A-E) and hypithermia (HT; F-J) groups at sham, 6 hours, 12 hours, 1 day, and 2 days after asphyxial cardiac arrest (ACA). Few toluidine blue positive mast cells were observed in the NT/sham and HT/sham groups. Numbers of mast cells (arrows) were increased after ACA, with a peak in number 1 day post-ACA in the NT/ACA group. In the HT/ ACA group, the change in number of mast cells over time was similar to that observed in the NT/ACA group, but there were significantly fewer mast cells than in the corresponding NT/ACA group. CV: central vein. Scale bar=50 µm. (K) Mean numbers of toluidine blue positive mast cells (n=7 at each time; aP<0.05, significantly different from NT/sham group; bP<0.05, significantly different from the previous timepoint group; cP<0.05, significantly different from NT/ACA group). Bars indicate mean±standard error of the mean.
|
Mast cells in the NT/sham group were also rarely shown around portal areas (Figure 1F). In the HT/ACA group, the number of mast cells at 6- and 12-hour post-ACA was similar to that in the NT/ACA group (Figure 1G, H, and K). At 1 day post-ACA, although the number of mast cells in the HT/ACA group was significantly increased relative to the control group, there were still significantly fewer mast cells (about 55.2%) than observed in the NT/ACA group (Figure 1I and K). At day 2 post-ACA, there were significantly fewer mast cells in the HT/ACA group than in the NT/ACA group (~60.5% of the mast cell numbers observed in the NT/ACA group) (Figure 1J and K).
DHE Fluorescence
ROS were detected using the fluorescent dye DHE (Figure 2). DHE fluorescence in the liver was very weak in the NT/sham (Figure 2A) and normal groups (data not shown). In the NT/ACA group, DHE fluorescence was significantly increased (about 173% of the NT/sham group) at 6 hours after ACA (Figure 2B and K). DHE fluorescence increased progressively and peaked at 1 day (about 318% of the NT/sham group) after ACA (Figure 2D and K). At 2 days after ACA, DHE fluorescence was slightly reduced (about 290 % of the NT/sham group) (Figure 2E and K).
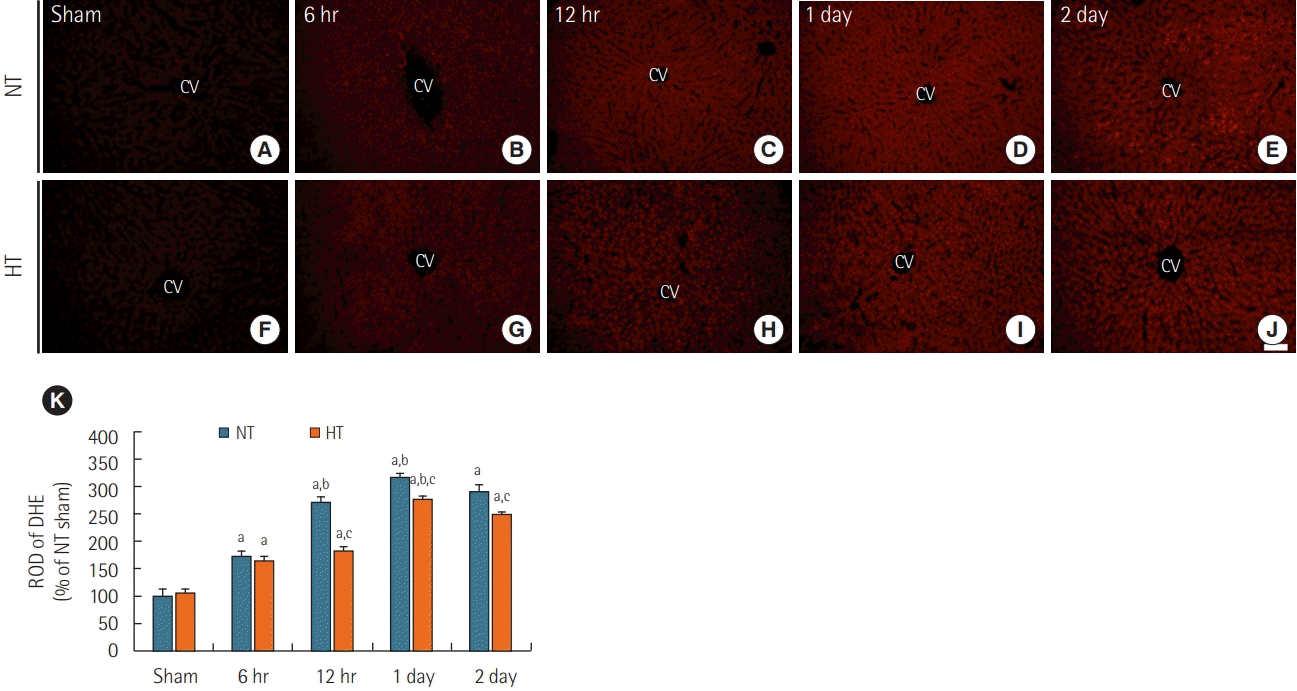 | Figure 2.Dihydroethidium (DHE) fluorescence staining in liver sections of normothermia (NT; A-E) and hypithermia (HT; F-J) groups at sham, 6 hours, 12 hours, 1 day, and 2 days after asphyxial cardiac arrest (ACA). Very weak DHE fluorescence was observed in the NT/sham and HT/sham groups. DHE fluorescence in both NT/ACA and HT/ACA groups gradually increased over time, but DHE fluorescence in the HT/ ACA group was lower than that in the NT/ACA group. CV: central vein. Scale bar=50 µm. (K) Relative optical density (ROD) of DHE fluorescence in all groups (n=7 at each time after ACA, aP<0.05, significantly different from NT/sham group; bP<0.05; significantly different from previous time-point group; cP<0.05, significantly different from NT/ACA group). Bars indicate means±standard error of the mean.
|
In the HT/sham group, DHE fluorescence was barely observed in the liver, like in the NT/sham group (Figure 2F and K). In the HT/ACA group, DHE fluorescence changed in a manner similar to that observed in the NT/ACA group; however, DHE fluorescence was significantly lower than in the NT/ACA group; the ROD of DHE fluorescence was about 94.5% at 6 hours, 66.8% at 12 hours, 87.4% at 1 day, and 86.2% at 2 days after ACA compared to that in the corresponding NT/ACA groups (Figure 2G-K).
SOD1 Immunoreactivity
SOD1 immunoreactivity was readily detected in the livers of the NT/sham group (Figure 3A) and normal group (data not shown). SOD1 immunoreactivity in the NT/ACA group was significantly lower than that in the NT/sham group at all times after ACA (Figure 3B-E), with ROD values of 43.4% at 6 hours, 62.5% at 12 hours, 30.8% at 1 day, and 36.2% at 2 days after ACA in comparison to values in the NT/sham group (Figure 3K).
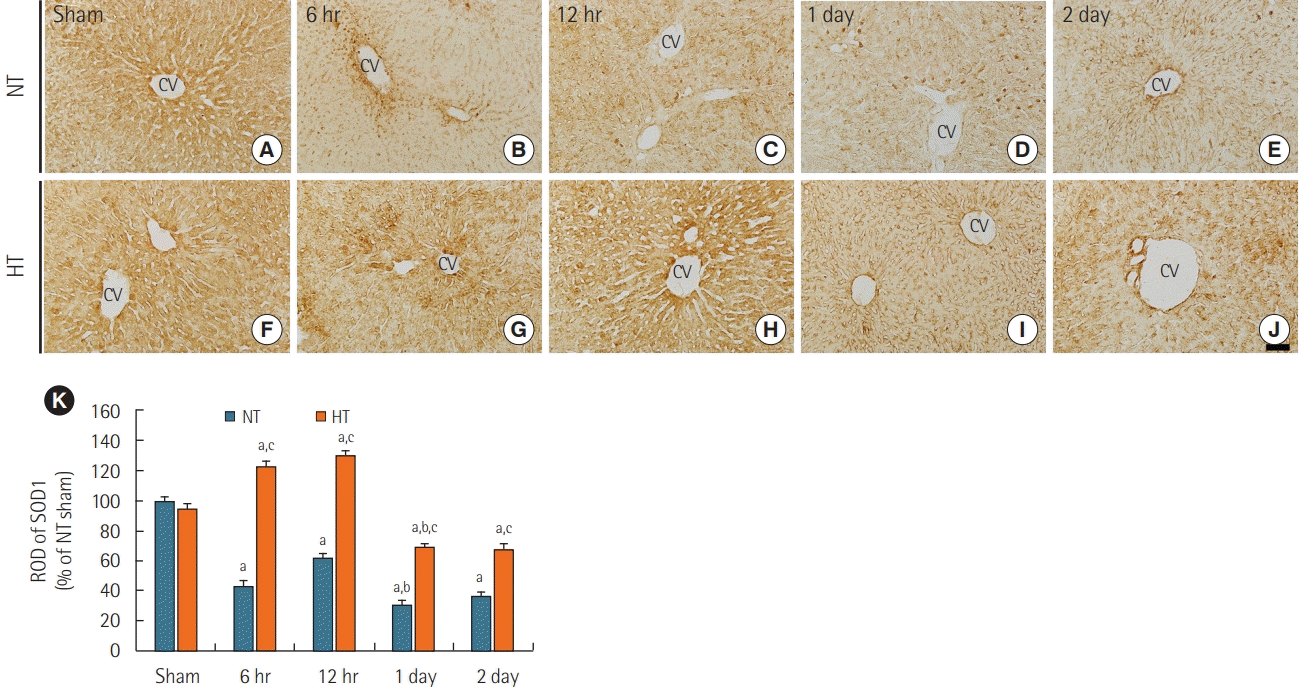 | Figure 3.Immunohistochemical staining for superoxide dismutase 1 (SOD1) in liver sections of normothermia (NT; A-E) and hypithermia (HT; F-J) groups at sham, 6 hours, 12 hours, 1 day, and 2 days after asphyxial cardiac arrest (ACA). SOD1 immunoreactivity decreased after 6 hours post-ACA in the NT/ACA group. SOD1 immunoreactivity was significantly higher in the HT/ACA group than in the NT/ACA group at all times after ACA. CV: central vein. Scale bar=50 µm. (K) Relative optical density (ROD) of SOD1 immunoreactivity in all groups (n=7 at each time after ACA, aP<0.05, significantly different from NT/sham group; bP<0.05, significantly different from previous time-point group; cP<0.05, significantly different from NT/ACA group). Bars indicate means±standard error of the mean.
|
SOD1 immunoreactivity in the HT/sham group was similar to that in the NT/sham group (Figure 3F). In the HT/ACA group, SOD1 immunoreactivity was significantly increased at 6 hours (about 123% of the NT/sham group) and 12 hours (about 130% of the NT/sham group) after ACA (Figure 3G, H, and K). SOD1 immunoreactivity decreased thereafter and was about 69% of the NT/sham group at 1 day and 68% of the NT/sham group at 2 days after ACA (Figure 3I-K). However, SOD1 immunoreactivity in the HT/ACA group was significantly higher at all times after ACA compared with that in the NT/ACA group (286.7% at 6 hours, 209.8% at 12 hours, 226.1% at 1 day, and 188.8% at 2 days after ACA relative to the corresponding NT/ACA group values, respectively) (Figure 3K).
SOD2 Immunoreactivity
Like SOD1 immunoreactivity in the NT/sham group, SOD2 immunoreactivity was readily detectable in the livers of the NT/sham group (Figure 4A). In the NT/ACA group, SOD2 immunoreactivity was markedly decreased by ACA with ROD values of 29.5% at 6 hours, 34.3% at 12 hours, 47.2% at 1 day, and 38.6% at 2 days compared to the corresponding time points in the NT/sham group (Figure 4B-E, and K).
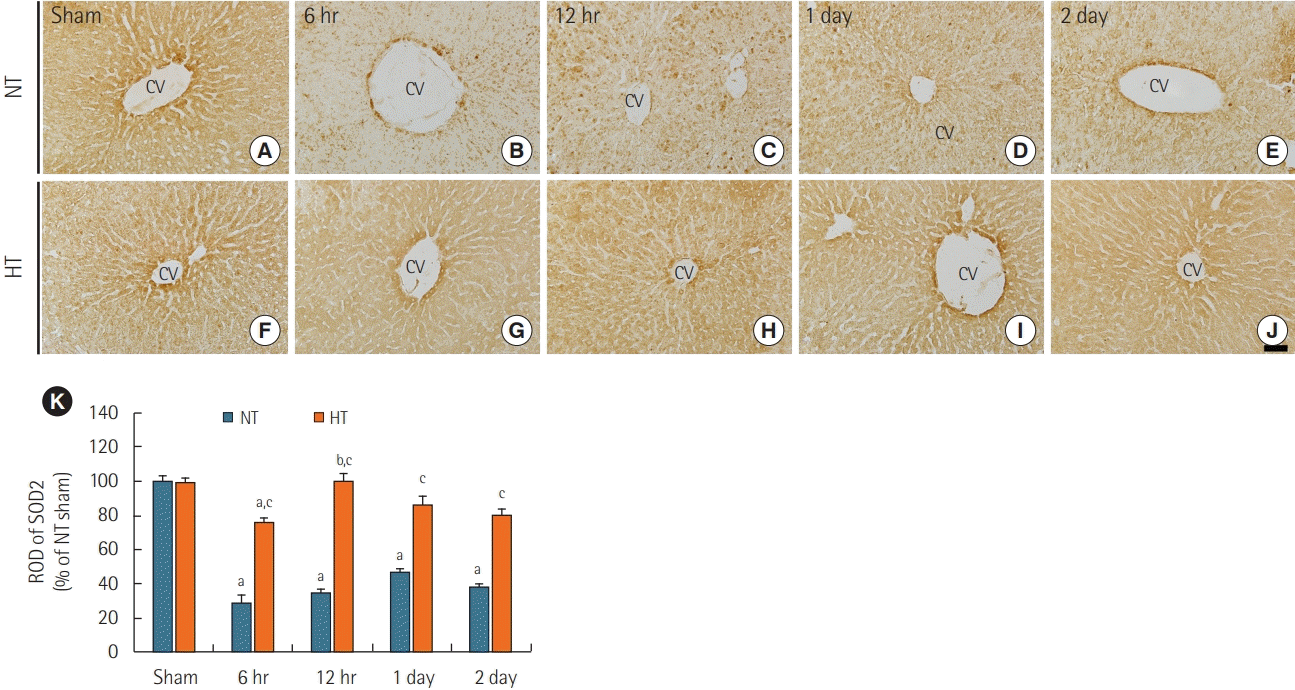 | Figure 4.Immunohistochemical staining for superoxide dismutase 2 (SOD2) in liver sections from the normothermia (NT; A-E) and hypithermia (HT; F-J) groups at sham, 6 hours, 12 hours, 1 day, and 2 days after asphyxial cardiac arrest (ACA). SOD2 immunoreactivity in the NT/ ACA group was markedly decreased 6 hours after ACA. In the HT/ACA group, SOD2 immunoreactivity did not change significantly after ACA. CV: central vein. Scale bar=50 µm. (F) Relative optical density (ROD) of SOD2 immunoreactivity in all groups (n=7 at each time after ACA, aP<0.05, significantly different from NT/sham group; bP<0.05, significantly different from previous time-point group; cP<0.05, significantly different from NT/ACA group). Bars indicate means±standard error of the mean.
|
SOD2 immunoreactivity in the HT/sham group was similar to that in the NT/sham group (Figure 4F). In the HT/ACA group, there was no significant change in SOD2 immunoreactivity compared to that in the NT/sham group (Figure 4G-J), but ROD values were significantly higher than those in the corresponding NT/ACA groups (about 262.1% at 6 hours, 290.9% at 12 hours, 182.9% at 1 day and 210.5% 2 days after ACA) (Figure 4K).
Go to :
DISCUSSION
Hypothermia treatment has been shown to prevent injuries to major organs such as the brain, kidney, and liver in experimental animals [17,18]. This study investigated the protective effects of hypothermia against I/R-induced inflammation and oxidative stress in the rat liver following 5 minutes of ACA. In short, the number of mast cells was significantly attenuated and oxidative stress was profoundly decreased in hypothermic rats compared to normothermic rats subjected to ACA.
Mast cells play key roles in the inflammatory process. When they are activated, they can release mediators in a piecemeal or anaphylactic manner from storage granules into the local microenvironment [30,31]. Toluidine blue staining is a commonly used method to count total mast cells in organs, including the liver [32]. In our current study, we used toluidine blue staining to detect mast cell infiltration in the liver and found that the number of mast cells was markedly increased in the NT/ACA group relative to the NT/sham group but was significantly reduced in the HT/ACA group relative to the NT/ACA group. I/R injury elicits activation of the inflammatory response [33]. Namely, mast cells promote I/R injury in many organs through their degranulation [34-36]. Mast cells release diverse active mediators such as histamine [37], tumor necrotic factor alpha, and platelet activating factor [38] following I/R, which regulate the inflammatory response following I/R injury [33,39]. Mast cell degranulation is induced directly by ROS, and contributes to the progression of I/R injury [40]. Some studies have demonstrated that inhibition of mast cell activation reduces the effect of the inflammatory response and attenuates liver damage caused by I/R insult [32,33,40]. In our current study, we showed that mast cells increased in number in the NT/ACA group after 5 minutes of ACA; however, in the HT/ACA group, the number of mast cells was significantly reduced compared to that in the NT/ACA group. This finding indicates that hypothermic treatment can attenuate I/R-induced liver inflammation thorough inhibition of the mast cell-mediated inflammatory response.
Intracellular superoxide anions (O2–) can be detected using DHE, a redox-sensitive probe [41]. Superoxide anion reacts with other molecules to form ROS like hydrogen peroxide (H2O2), the hydroxyl radical (•OH), and reactive nitrogen species [42]. I/R insult increases the production of ROS, which elicits oxidative stress [43]. In a porcine model of CA, hypothermia reduced reactive oxygen metabolite levels compared to those under normothermia [18]. In post-CA patients, plasma reactive oxygen metabolite levels decreased during hypothermia treatment (33°C) [19]. Additionally, in rats, acute liver ischemia induced oxidative stress, which was attenuated by hypothermia [4]. We demonstrated a gradual increase in superoxide anion radical production in the rat liver over time in the NT/ACA group, but production of this radical was significantly suppressed in the HT/ACA group. This finding indicates that therapeutic hypothermia can effectively reduce oxidative stress after hepatic I/R.
We also examined the immunoreactivities of SOD1 and SOD2 and found that levels of these enzymes were significantly decreased in the NT/ACA group compared to the NT/sham group. SOD activity has been shown to decrease in the liver, lung, and ileum in rat models of I/R injury [44-46]. SOD levels decrease after I/R [6], and the consumption of SODs due to ROS overproduction causes cell damage or death [12]. We evaluated whether hypothermia treatment was associated with an imbalance antioxidants in the rat liver by examining the expression of antioxidant enzymes (SOD1 and SOD2) by immunohistochemistry and found that immunoreactivities of SOD1 and SOD2 were much higher in the HT/ACA group than in the NT/ACA group.
In liver I/R injury in mice, hypothermia significantly enhanced the expression of SOD1 (cytoplasmic SOD) and SOD2 (mitochondrial SOD) [2]. In addition, intravenous treatment with SOD reduced apoptotic cell death and oxygen free radical production after I/R in rats [6]. Furthermore, I/R injury in skeletal muscle in rats was associated with decreased SOD activity, which was restored by hypothermia treatment [46]. In a pig model of CA, hypothermia treatment increased SOD2 expression in the frontal cortex after CA-induced I/R injury [17]. Consistent with these findings, hypothermic treatment has been reported to offer protection by enhancing the endogenous defense system and increasing the expression of antioxidant enzymes [47]. In addition, consumption of endogenous antioxidants due to the release of ROS causes apoptotic and necrotic cell death [12] and hypothermia reduces apoptosis [48] and necrotic damage at the cellular level by reducing ROS accumulation [49]. Increased immunoreactivities of liver SOD1 and SOD2 in the HT/ACA group suggest that hypothermia treatment protects against liver inflammation by increasing the expression of antioxidant enzymes that then scavenge oxygen free radicals after I/R. Namely, SOD1 and SOD2 as the main oxygen radical scavengers convert superoxide anion radicals thereby protecting cells against I/R-induced liver inflammation [2].
In short, we demonstrated that hypothermia treatment decreased the ACA-induced increase in number of mast cells and ROS production, and enhanced the expression of SODs after ACA. Therefore, we suggest that therapeutic hypothermia has antioxidant and anti-inflammatory effects in the liver following ACA. Further studies are needed to better understand the protective effect of hypothermia after I/R and to determine whether hypothermia treatment is beneficial for transplantation of organs prone to I/R injury.
Our study has some limitations that should be considered when performing follow-up studies. We did not investigate indicators of liver damage such as aspartate aminotransferase and alanine aminotransferase. In addition, we focused only on SOD1 and SOD2 as antioxidants enzymes. Therefore, in the future, levels of liver damage markers in the blood should be assessed and the expression of antioxidants enzymes such as CAT and GSH should also be investigated.
Go to :
KEY MESSAGES
▪ Hypothermia reduces superoxide anion production in the liver after asphyxial cardiac arrest (ACA).
▪ Hypothermia increases the expression of antioxidant enzymes in the liver after ACA.
▪ Antioxidative effects of hypothermia can reduce the ACA-induced increase in mast cell numbers in the liver.
Go to :
 XML Download
XML Download