

 PDF
PDF Citation
Citation Print
Print



Introduction
Acute respiratory distress syndrome (ARDS) is a refractory hypoxemic respiratory failure with bilateral lung infiltrates, in the absence of left atrial hypertension or evidence of volume overload [1,2].
The ARDS is associated with high mortality rates, which vary widely from 30% to 70% in several reports [3,4]. Although the annual ARDS mortality rates have shown improvement in several studies [5,6], ARDS remains a life-threatening disease with high mortality.
One of major pathophysiologic mechanisms in the early phase of ARDS is known to be endothelial injury and alveolar epithelial damage from proinflammatory cascade [7] , leading to the process of proliferation and fibrosis [8]. Inflammatory biomarkers such as tumor necrosis factor-alpha, interleukin 6, platelet activating factor are known to involve in ARDS pathogenesis [9-11]. In the advanced stage, activated alveolar fibrocytes, fibroblasts and myofibroblasts may drive fibroproliferation, although the process remains unclear [12]. These inflammatory cytokines and increased pulmonary fibrosis predict unfavorable outcome and higher mortality [13,14]. Therefore, the anti-inflammatory therapy or prevention of pulmonary fibrosis in ARDS could be targets of therapy to improve survival.
Angiotensin II exerts proinflammatory responses through activating nuclear factor-κB in monocytes [15]. The renin-angiotensin system (RAS) inhibitors, such as angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB) reduces nuclear factor-κB activation and lipopolysaccharide induced lung neutrophil recruitment [15-17]. Rats with acute lung injury showed lower lung injury score or alveolar collagen deposit when treated with captopril and they suggested inhibition of ACE could offer protective effects on acute lung injury [18-21]. The mechanism is supposed that captopril attenuates lung fibrosis by abrogating apoptosis in lung epithelial cells.
ACE inhibitor is also well known to have a protective effect against fibrosis and remodeling, notably in the liver, kidneys and heart [22-24]. However, there was little evidence that ACE inhibitor played a protective role in human ARDS.
We hypothesized that ACE inhibitor could provide beneficial effects for ARDS patients by protecting them from prolonged inflammation and fibrosis. Therefore, we conducted this study to determine whether there is a difference in clinical outcomes including mortality rates in ARDS patients according to the use of ACE inhibitor or ARB.
Materials and Methods
1) Patients population
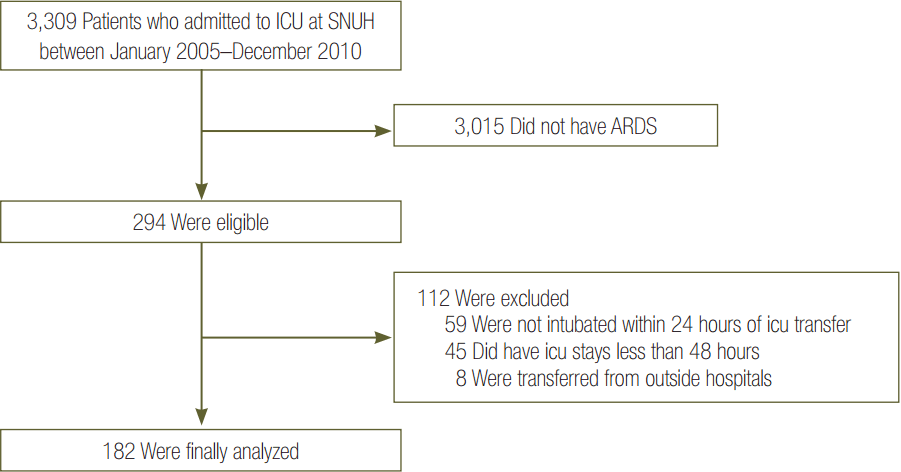
We retrospectively reviewed the medical records and radiographs of patients admitted to the medical intensive care unit (MICU) at a tertiary care hospital for mechanical ventilation support between January 2005 and December 2010. Eligibility criteria were that patients were: (1) over 20 years of age, and (2) fulfilled the Berlin definition: acute onset within 1 week of a known clinical insult or new or worsening respiratory symptoms; bilateral opacities not fully explained by effusions, lobar/lung collapse, or nodules; respiratory failure not fully explained by cardiac failure or fluid overload; arterial hypoxemia with PaO2/FiO2 lower than 300 (positive end-expiratory pressure [PEEP] or continuous positive airway pressure, ≥5 cmH2O) [25]. We excluded patients who were not intubated within 24 hours of ICU transfer or did not stay in the ICU for more than 48 hours following admission. Patients who were intubated and transferred from outside were also excluded.
2) Study design and data collection
Patients were classified into two groups according to whether or not they took ACE inhibitor/ARB after ICU admission (RAS inhibitor group and non-RAS inhibitor group, respectively). The study was approved by the institutional review board and ethics committee (No. H-1206-090-414). No consent from the participants was needed.
Demographic data (age, gender), risk factors for ARDS and etiology of ARDS were evaluated. We also calculated the PaO2/FiO2 ratio, Sequential Organ Failure Assessment (SOFA) score (at admission, 48 hours and 96 hours after admission) and Acute Physiology and Chronic Health Evaluation (APACHE) II score (within 24 hours after admission). Medication histories were reviewed to find the patients who took ACE inhibitor or ARB after ICU admission, and to collect data about the generic name, dose and duration of medication.
The primary outcome was ICU mortality, and secondary outcomes we measured by length of stay in ICU, ICU readmission rate, reintubation rate, radiologic improvement (decreased consolidation or infiltrates on chest radiography at ICU discharge compared to ICU admission) weaning failure rate (rate of reintubation within 48 hours after extubation) and 28- and 90-day in-hospital mortality.
3) Statistical analysis
Quantitative variables are expressed as mean ± standard deviation, and qualitative variables are expressed as numbers and percentages. Differences between independent groups were assessed by paired t-tests for quantitative data and chi-square tests for qualitative variables. We used propensity score matching to reduce the bias caused by confounding variables. The propensity score covariates included age, gender, preexisting disease (hypertension, diabetes, coronary artery disease, chronic obstructive pulmonary disease, tuberculosis, chronic liver disease, chronic kidney disease, and malignancy) and clinical disease severity scores (SOFA score and APACHE II score). Patients with similar propensity scores in each group were matched with 1 to 1 ratio. Outcomes were compared in both unmatched and matched cohort. In the ICU and hospital, mortalities were estimated with the Kaplan-Meier analysis and log-rank test using propensity score matching analysis. All statistical analyses were performed using Stata version 13.0 (Stata Corp., College Station, TX, USA). A P-value <0.05 was considered to indicate a significant difference.
Results
We reviewed the records of 3,309 patients admitted to the ICU between January 2005 and December 2010. Of the 294 patients who satisfied the inclusion criteria, 112 patients were excluded, leaving 182 patients for the final analysis. After the propensity score matching, 34 patients in each group were analyzed (Figure 1).
Of the 182 patients, 134 (74%) were male and the mean age was 64.1 ± 13.7 years. Thirty-seven patients received ACE inhibitor and ARB during their ICU stay. Table 1 presents the patients’ demographic characteristics and history of underlying disease. History of hypertension and chronic kidney disease were significantly higher in the RAS inhibitor group. Though history of malignancy was higher in patients in the non-RAS inhibitor group, patients with lung cancer were higher in RAS inhibitor group. (P = 0.590) No difference in the proportion of direct lung injury was observed between the two groups.
Eighty-nine patients (48.9%) were classified as severe ARDS according to the Berlin definition. Proportion of patients with severe ARDS was higher in the RAS inhibitor group (54.3%) than non-RAS inhibitor group (47.6%) without statistical significance (P = 0.431). Proportions of patients with mild ARDS were 4.8% and 8.6%, those with moderate ARDS were 47.6% and 37.1% in non-RAS inhibitor group and RAS inhibitor group, respectively.
We analyzed the PaO2/FiO2 ratio (initial, 24 hours, 72 hours), SOFA score (initial, 48 hours, 96 hours), and APACHE II score and found no differences between the two groups.
The average PEEP levels were about 6.9 cmH2O in both groups (P = 0.681). Tidal volumes were also similar, those values were 7.4 ± 1.9 ml/kg and 7.5 ± 1.9 ml/kg, respectively in non-RAS inhibitor group and RAS inhibitor group (P = 0.774). We constructed the propensity score matched cohort, which consisted of 34 patients in each group. Demographic characteristics, comorbidities and mechanical ventilator parameters were similar between two groups in the propensity score matched cohort.
About 60% of the patients in the RAS inhibitor group had taken ACE inhibitor or ARB within 1 month before MICU admission (Table 2). In the RAS inhibitor group, 10 patients had been given ACE inhibitor and 27 patients were given ARB.
For the patients receiving ACE inhibitor/ARB, ICU mortality was 45.9% (17 of 37 patients), which was lower than the 58.9% mortality rate of patients without medication, though not significantly (P = 0.166) (Table 3). Inhospital mortality was also higher in the non-RAS inhibitor group and the day 28 mortality in non-RAS inhibitor group was almost twice as high without statistical significance (18.9% vs. 35.2%, P = 0.058). In the propensity score matched cohort, in hospital mortality at day 28 was also higher in non-RAS inhibitor group (29.4%) than RAS inhibitor group (8.8%, P = 0.031). However, other parameters of outcome including in hospital mortality at day 90 showed no significant differences between two groups.
ICU-free day at 28 days were about 5 days, similar in both groups (P = 0.822). ICU-free day at 90 days was longer in RAS inhibitor group (mean, 34.2 days) than non-RAS inhibitor group (mean, 29.4 days; P = 0.481). There was also no significant difference in hospital-free day at 90 days and ventilator-free day at 28 days, between two groups.
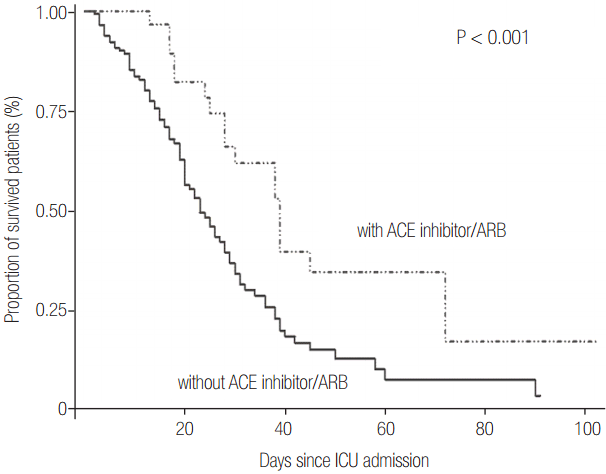
The Kaplan-Meier estimate of survival showed a higher mortality rate in non-RAS inhibitor patients (P < 0.001) (Figure 2). Survival analysis after adjustment for propensity score showed similar results in each group (P = 0.002).
Discussion
Although many studies have been conducted to establish pharmacologic management of patients with ARDS, there is no proven pharmacologic therapy to improve the survival and prognosis of ARDS patients [26]. Clinical trials using well-known anti-inflammatory agents, including corticosteroids, 3-hydroxy-3-methylglutaryl-coenzyme-reductase inhibitors, heparins and aspirin, have not demonstrated clear clinical benefits in ARDS [27-30].
Interestingly, several studies conducted to support anti-inflammatory effect of ACE inhibitor in vascular disease [31-33] and also showed protective effect against pneumonia [34,35]. In addition, some animal models have revealed that administration of ACE inhibitor or ARB inhibits fibrosis in the kidneys, heart and liver [36-39]. We therefore tried to evaluate the effect of anti-inflammatory and antifibrotic properties of ACE inhibitor and ARB on outcomes in patients with ARDS.
In this study, radiologic improvement was more frequently detected in the RAS inhibitor group, although this difference was not significant. The mean PaO2/FiO2 ratio on admission was lower in the RAS inhibitor group than in the non-RAS inhibitor group (103.1 ± 51.7 vs. 112.2 ± 45.4, P = 0.297). However, the ratio increased over time than non-RAS inhibitor group (187.8 ± 113.6 vs. 173.1 ± 90.9, P = 0.408). Clinical improvement in the late phase in regards to chest radiology and oxygenation, might reflect lower degrees in pulmonary sequelae including fibrosis in RAS inhibitor group.
An interesting finding was that the ACE inhibitor group showed greater ICU stay duration and length of mechanical ventilation. Previous studies that conducted survival analysis suggested that surviving patients stayed longer in the ICU [31,40,41]. This could explain the differences between the two groups. Considering that no differences in ICU-free day or ventilator-free day, longer duration of ICU or mechanical ventilation could not be affirmed as poor clinical outcome.
More patients in the RAS inhibitor group were transferred to general wards, supported by portable bi-level positive airway pressure ventilators (18.9% vs. 5.5%, P = 0.008). Considering the similar results in terms of PaO2/FiO2 ratio and radiologic improvement on discharge, higher dependence on the bi-level positive airway pressure ventilator might be mainly caused by muscle weakness [42].
The mortality rates, both in the ICU and in the hospital, were not different in both groups. The overall mortality rate was high compared to other cohorts, which estimated mortality rates ranged from 28% to 58% [31,43,44]. It might reflect high disease severity considering high APACHE II score and high proportion of cancer patients in our study [45]. Among the patients who died, the duration of ICU stay was longer (P = 0.001) in the RAS inhibitor group than in the non-RAS inhibitor group, which might explain the lower 28-day mortality rates. Survival analysis showed significantly better survival rates in the RAS inhibitor group. We adjusted confounding factors, including disease severity, using the propensity score, and found the same results as above. The likelihood of survival of patients with ARDS could be improved by the administration of RAS inhibitor.
This study has several limitations. First, ARDS is a complicated syndrome, and it is difficult to distinguish ARDS from other diseases that might cause radiologic abnormality and respiratory failure. However, we did our best to overcome the weakness of the retrospective design. We reviewed all medical records, including echocardiography results, daily body weight changes and vital sign changes, to evaluate volume status. Records of physical examination and assessment by medical attendance were also reviewed to exclude other diseases. Second, ACE inhibitors or ARB administered to patients did not have uniform component or dosage. However, for the treatment of hypertension, ACE inhibitor and ARB showed similar efficacy in lowering blood pressure [46,47]. Though the mechanisms of action between ACE inhibitor and ARB are different, there was no relevant difference in clinical outcomes for renal protection [48]. Further researches are needed to compare efficacy in anti-inflammatory effect. Third, the sample size was relatively small, and a considerable number of patients had many comorbidities. These confounding factors might have influenced the outcome [49].
In conclusion, in this study we found that ACE inhibitor or ARB may have beneficial effect on ARDS patients. We need large prospective clinical study to validate this finding.
 XML Download
XML Download