

 PDF
PDF Citation
Citation Print
Print


Introduction
Septic shock is characterized by multiple cardiovascular abnormalities including absolute and relative hypovolemia, vascular tone depression, myocardial dysfunction, derangements in regional blood flow distribution and microcirculatory disorders. Septic shock is often associated with multiple organ failure, including mainly respiratory and renal failures. The degree of severity of each of these disorders is variable from patient to patient. Early and adequate hemodynamic support is crucial to prevent further worsening of organ dysfunction. Therefore, it is important to assess the degree of each component of the hemodynamic failure in order to select the appropriate therapeutic measures. Multiple hemodynamic variables are available to manage patients with septic shock. The main issue for the physician is to properly integrate these variables in order to optimize the therapy. Some of the hemodynamic variables are used to make or confirm the diagnosis of shock and others are used to guide therapy and to evaluate the response to it. In this review, we describe a wide variety of hemodynamic variables and we propose how to integrate them in a therapeutic strategy for septic shock patients.
What are the most relevant hemodynamic variables during resuscitation of septic shock?
1) Blood Lactate
Lactate is mainly released by the skeletal muscles, the adipose tissue and the brain and in a smaller proportion by the lung and the heart. Daily, 20 mmol/kg of lactate is produced, released in the blood stream and transported to the liver and the kidney, where it is subsequently metabolized in two main energy (ATP)-producing processes: glycolysis and oxidative phosphorylation. Lactate arises from the metabolism of glucose. Glycolysis metabolizes glucose to pyruvate in the cellular cytosol. Further metabolism of pyruvate follows one of two routes. First, pyruvate enters mitochondria and is converted to acetyl-coenzyme A by pyruvate dehydrogenase, which enters the Krebs’s cycle allowing generation of 38 ATP molecules for each molecule of metabolized glucose. The second route for pyruvate is conversion to lactate in the cytosol. This reaction is bi-directionally catalyzed by lactate dehydrogenase, resulting in a normal lactate/pyruvate ratio of approximately 10.
Development of hyperlactatemia in sepsis is multifactorial. It can be due to an overproduction of lactate via either anaerobic or aerobic metabolism or to a decrease in its clearance. During sepsis, impaired ability of the tissues to extract oxygen and endothelial inflammatory processes result in microcirculatory dysfunction so that regional DO2 is not matched with oxygen demand and tissue hypoxia develops.[1] When sufficient oxygen is not available, the Krebs’s cycle cannot metabolize pyruvate so that lactate is generated. In addition, mitochondrial dysfunction may occur during sepsis so that, even in tissues with adequate oxygenation, pyruvate is shunted toward lactate production. In addition, the resting metabolic rate can be increased during sepsis leading to increased metabolism of glucose.[2] Glycolytic flux can exceed the capacity of pyruvate dehydrogenase to catalyze conversion of pyruvate into acetyl coenzyme A. Therefore, pyruvate is converted to lactate by lactate dehydrogenase. Finally, β2 adrenergic stimulation can activate the sodium-potassium-adenosine triphosphatase (Na-K-ATPase) activity and this increased the glycolytic flux.[3] These results are supported by the observation that lactate production is reduced by β2-receptors antagonists such as esmolol and by Na/K-ATPase inhibitors (ouabain) in septic shock or when glycolysis is induced by epinephrine.[4] Finally, hyperlactatemia might also be the result of impaired lactate clearance during sepsis. [5] This may become an important issue in patients with significant pre-existing or new hepatic dysfunction.
2) Arterial blood pressure
Arterial catheterization (radial or femoral artery) is recommended as soon as possible during acute circulatory failure to monitor the arterial pressure waveform in real-time and also to easily sample arterial blood.[6,7] This provides direct measurements of systolic arterial pressure and diastolic arterial pressure (DAP) and from these two values, estimation of mean arterial pressure (MAP) and of pulse pressure (PP). Monitoring these four pressures provides important information on the hemodynamic status and the cardiac function.
(1) Mean arterial pressure
The MAP is considered as the upstream pressure for perfusion of most vital organs such as the brain and the kidney. As a result, when MAP falls below the lower limit of autoregulation, regional blood flow becomes linearly dependent upon MAP. In some pathological settings, and in specific vascular areas, MAP underestimates the true perfusion pressure, because of marked increases in the downstream venous pressure or in the extravascular pressure at the outflow level. There is no universally admitted MAP threshold ensuring that blood flow is independent of arterial blood pressure in most vital organs. Indeed, the critical level of MAP probably differs among organs and depends on numerous factors including age, previous history of hypertension, neuro-vegetative state and vasoactive therapy. However, in septic shock, current resuscitation guidelines recommend to initially achieve MAP ≥ 65 mmHg, in order to avoid additional organ hypoperfusion[6,8] and suggest a higher value in patients having prior chronic hypertension, a pathological state characterized by a rightward shift of the autoregulation curve. Recently, a multicenter randomized open-label trial was conducted to compare two MAP targets in patients with septic shock: 65-70 mmHg and 80-85 mmHg[9] There was no difference between the two groups in 28-day mortality rates.[9] There was no difference either in the overall rate of serious adverse events, except a slightly higher rate of atrial fibrillation in the high MAP target group.[9] In the predefined stratum of patients with chronic hypertension, those with a MAP target of 80-85 mmHg exhibited a more pronounced improvement in renal function and fewer requirements for renal replacement therapy.[9] This study thus suggests that in patients with history of chronic hypertension, a MAP between 80-85 mmHg is a reasonable target. A MAP target higher than 65 mmHg should also be considered in cases of high central venous pressure (CVP), which reflects a high outflow venous pressure within organs. Indeed, in such cases, it could be important either to increase the MAP or to decrease CVP in order to maintain an adequate driving pressure for organ perfusion. Similarly, in intraabdominal hypertension syndromes, the increased intraabdominal pressure can represent the outflow pressure of intra-abdominal organs, necessitating then a higher than normal MAP target to preserve their adequate perfusion unless therapeutic reduction of the intra-abdominal pressure is feasible.
(2) Diastolic arterial pressure
The DAP is also important to monitor since it is the driving pressure for the left ventricular perfusion. In addition, DAP is a marker of arterial tone. It is also influenced by arterial compliance and heart rate. A low DAP can thus exist in cases of decreased vasomotor tone (e.g. sepsis), reduced arterial compliance and bradycardia. Therefore, in case of sepsis with tachycardia, a low DAP (i.e. < 40 mmHg) is an indicator of a profoundly depressed arterial tone.
(3) Pulse pressure
At the level of aorta, PP is dependent on stroke volume and arterial stiffness. Due to the pulse wave amplification (PWA) phenomenon, which increases the peripheral PP compared to the aortic PP, the relation between peripheral PP and stroke volume is weaker except in old patients or in those with chronic hypertension, diabetes or arteriosclerosis, in whom the PWA is minimal. In those patients, PP should be higher than normal (60-80 mmHg) if stroke volume is normal due of the increased arterial stiffness. Thus, a lower than normal PP (30-40 mmHg) in such patients means than stroke volume is low.
3) The mixed venous blood oxygen saturation (SvO2) and the central venous oxygen saturation (ScvO2)
The SvO2, which is measured in the mixed venous blood is an integrative variable that reflects the global balance between arterial oxygen delivery (DO2) and the whole-body oxygen consumption (VO2) since cardiac output (CO), hemoglobin concentration (Hb) and arterial blood oxygen saturation (SaO2) are the key determinants of DO2. Indeed, SvO2 is related to arterial SaO2, VO2, CO and Hb according to the formula derived from the Fick equation applied to oxygen: SvO2 = SaO2 - (VO2 / [CO × Hb × 13.4]). In healthy subjects at rest with normal SaO2 and Hb, the value of SvO2 ranges from 70 to 75%. SvO2 can be considered as a marker of oxygen extraction. Indeed, the whole-body oxygen extraction ratio (OER) is defined as the ratio of the whole-body VO2 to the wholebody DO2: OER = VO2/DO2. As DO2 equals approximately CO × Hb × 13.4, then SvO2 equals SaO2 - (VO2/DO2). When SaO2 is close to 1 (100%), SvO2 can thus be used to estimate OER: SvO2 = 1 - OER.
By considering that SvO2 reflects OER, SvO2 changes can be interpreted as follows:
A decrease in SvO2 means that OER has increased to make the whole-body VO2 matched with the wholebody oxygen demand. This increase in OER can be triggered by either a drop in DO2, or an increase in oxygen demand, or both. If, in spite of this normal adaptation, VO2 does not match oxygen demand, tissue hypoxia and anaerobic metabolism appear.[10] However, no precise value of SvO2, can be defined as the “critical” SvO2 below which tissue hypoxia occurs.[10]
Conversely, an increase in SvO2 means that OER has decreased as a consequence of either an increase in DO2 with maintained VO2 or a decrease in VO2 with maintained or increased DO2. This latter situation is typically encountered in hyperdynamic septic shock, where the decrease in VO2 (despite a high whole-body oxygen demand) does not result from a reduced DO2, but rather from impairment of the O2 extraction capabilities. These points underline the fact that: 1) SvO2 cannot be a reliable marker of the adequacy between DO2 and global oxygen demand in shock states, where VO2 is lower than oxygen demand, and 2) interpretation of SvO2 can be misleading in hyperdynamic states.[11] Finally, it should be kept in mind that during resuscitation of shock states, increase in DO2 is expected to result in parallel increase in VO2 such that SvO2 does not change markedly unless a critical level of DO2 is reached. Above this critical level of DO2, a further increase in DO2 is associated with a marked increase in ScvO2 but no anymore change in VO2.[11]
Due to the decline of the use of the pulmonary artery catheter, ScvO2 has been proposed to replace SvO2, as a central venous catheter is recommended to be used in shock states.[6,7] Studies comparing SvO2 and ScvO2 generally found satisfactory results in terms of absolute values[12] as well as in terms of trends.[13] Confirmation of the interest of ScvO2 in the resuscitation of septic shock patients was brought by the Rivers et al’s study,[14] where a therapeutic strategy (called early goal-directed therapy) aimed at targeting a ScvO2 value > 70% during initial resuscitation was shown to result in a better outcome than standard care.[14] Although recent multicenter randomized clinical trials did not confirm these findings,[15] it must be emphasized that ScvO2 was already higher than the 70% target in the vast majority of the included patients so that these trials could not by nature showed any benefit of EGDT.
4) Mixed venous blood (or central venous blood) minus arterial blood carbon dioxide (CO2) pressures differences
The Fick equation applied to CO2 indicates that the CO2 excretion (equivalent to CO2 production (VCO2) in a steady-state) equals the product of CO by the difference between the CO2 content in the mixed venous blood and in the arterial blood. The normal relationship between CO2 content and CO2 pressure is almost linear over the usual physiological range of the CO2 contents. Thus, by substituting CO2 pressure for CO2 content, the mixed venous blood – arterial blood PCO2 difference (also called PCO2 gap) equals k × VCO2 / CO, where k is a factor defining the relationship between CO2 content and CO2 pressure.[16] The relationship between CO and CO2 pressure is explained by the CO2 stagnation phenomenon that increases the venous blood CO2 pressure relatively to the arterial blood CO2 pressure at the peripheral venous level. Thus, a low CO and hence a low efferent venous blood flow should decrease the CO2 clearance rate and should result in an increased PCO2 gap for a given VCO2. In normal conditions, PCO2 gap values range between 2 and 6 mmHg. Accordingly, in a clinical study performed in normolactatemic patients with low cardiac index at baseline, the increase in cardiac index from 1.6 to 2.2 L.min-1.m-2 was accompanied by a decrease in PCO2 gap from 9 to 5 mmHg, while VO2 (and thus probably VCO2) was unchanged.[17]
In patients with tissue hypoxia, two situations should be distinguished in function of the systemic blood flow. In case of tissue hypoxia with reduced systemic blood flow, PCO2 gap should increase, whereas in case of tissue hypoxia with maintained or high systemic blood flow, PCO2 gap should be normal.[18,19]. In the latter conditions, the high venous blood flow can easily remove the low amount of CO2 produced at the periphery.
In summary, regardless of the presence of tissue hypoxia, a normal PCO2 gap (≤ 6 mmHg) is expected if CO is normal or high, and a high PCO2 (> 6 mmHg) is expected if CO is too low. Thus, a normal PCO2 gap suggests that elevation of CO cannot be a priority in the therapeutic strategy, whereas a high PCO2 gap suggests that elevation of CO may be a good therapeutic option.
5) Dynamic variables and tests predicting fluid responsiveness
The concept of prediction of fluid responsiveness has emerged since it has been demonstrated that only 50% of patients with acute circulatory failure respond to fluid administration in terms of increase in CO[20] and that fluid overload is associated with increased mortality.[21] During the last fifteen years, a number of indices able to predict fluid responsiveness at the bedside have been developed. In patients receiving mechanical ventilation, indices based on heart-lung interactions and mainly the respiratory variation of surrogates of stroke volume have been proposed.[22] The general principle is that when the heart is preload responsive, the stroke volume - or a surrogate such as PP - varies markedly over the respiratory cycle.[22] Accordingly, pulse pressure variation (PPV) was found to predict responsiveness better than cardiac filling pressures in patients with septic shock. [23] A metaanalyse including more than 800 patients showed that the prediction of fluid responsiveness by PPV is excellent in various clinical conditions with a cut-off of 12%.[24] In other words, when PPV is > 12%, CO is likely to increase by more than 15% if fluid is infused (fluid responsiveness), whereas if PPV is < 12%, no increase in CO can be expected from fluid infusion (fluid unresponsiveness). Recent functional hemodynamic monitoring devices calculate PPV automatically from the arterial pressure curve recording and display its value in real-time on their screen. Other fluid responsiveness indices such as stroke volume variation (SVV) can also been calculated and displayed by some advanced hemodynamic monitors but they have no superiority over PPV,[25] which has the great advantage to be recorded from a simple arterial catheter. However, the applicability of PPV (or SVV) for predicting fluid responsiveness is limited in the intensive care unit since it cannot be used in cases of spontaneous breathing activity, cardiac arrhythmias, low tidal volume ventilation and low lung compliance.[26] Other tests such as passive leg raising (PLR)[27] reliably predict fluid responsiveness,[28] in particular in situations where PPV fails to do it.[29,30] Strict rules must be respected for the PLR being adequately interpreted. [27] Among them, the need of a real-time CO measurement must be emphasized. Real-time CO monitors using the arterial pressure waveform-based CO[31] as well as echocardiography (velocity-time integral)[32] are particularly suitable.
6) Transpulmonary thermodilution variables
Transpulmonary thermodilution systems have been developed in the last fifteen years and are currently recommended to be used in complex patients, especially in those with circulatory shock and associated ARDS.[6,7] They take advantage of providing extravascular lung water (EVLW) and pulmonary vascular permeability index (PVPI), which represent quantitative measures of the volume of pulmonary edema and of the degree of lung capillary leakage, respectively.[33] Values of EWLW > 10 mL/kg[34] and of PVPI > 3[35] are considered as abnormally increased. In addition to EVLW and PVPI, the transpulmonary thermodilution systems provide intermittent measurements of CO and global end-diastolic volume (a volumetric marker of cardiac preload) and continuous CO and PPV, using the pulse contour analysis method.
How can clinicians integrate the hemodynamic variables to manage patients with septic shock?
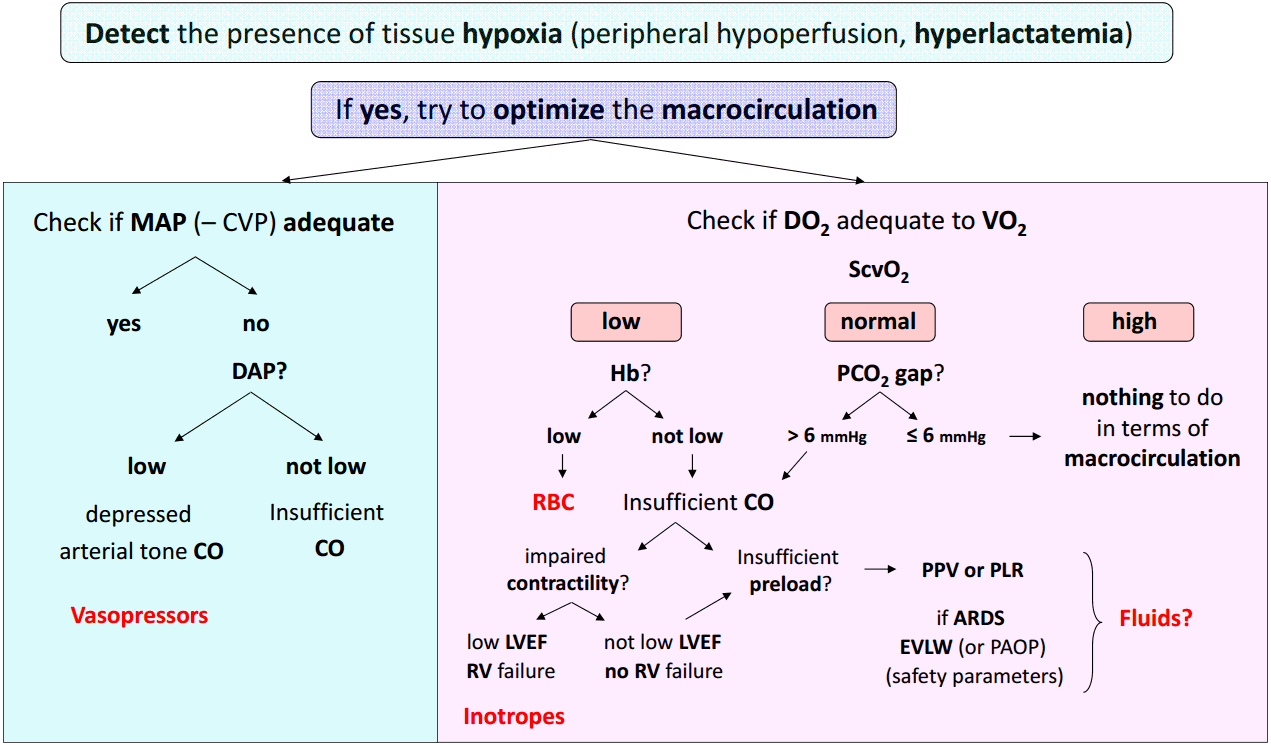
The Fig. 1 depicts a step-by-step therapeutic approach of septic shock integrating the main hemodynamic variables.
1) First step: detect acute circulatory failure and tissue hypoxia
Presence of hyperlactatemia along with clinical signs of peripheral hypoperfusion (mottling, increased capillary refill time) is fundamental to detect global tissue hypoxia. When present, hypotension is also a good indicator of circulatory failure, but a normal arterial blood pressure cannot rule out the diagnosis of shock at least in its early phase.[6]
As we mentioned above hyperlactatemia may have other origins than tissue hypoxia but in such a context, it is considered as an excellent quantitative marker of tissue hypoxia. The ideal tool to detect the presence of tissue hypoxia would be the lactate/pyruvate ratio (see above). If it is far above 10, this strongly suggests that hyperlactatemia is actually related to anaerobic metabolism and this should urgently prompt therapies aimed at reducing tissue hypoxia. If the lactate/pyruvate ratio is around 10, hyperlactatemia is probably due to accelerated aerobic metabolism and not to tissue hypoxia. Unfortunately in the routine practice, lactate but not pyruvate is measured so that the clinician should always interpret hyperlactatemia in function of the clinical context. In addition to the baseline blood lactate concentration, the blood lactate kinetics is important to assess. A decrease in blood lactate concentration with therapy is currently recognized as a valuable target for hemodynamic resuscitation in shock states.[36,37]
2) Second step: once the presence of tissue hypoxia is detected, check at the same time both the arterial blood pressure and the DO2/VO2 balance.
(1) Check arterial blood pressure
As mentioned above severe hypotension can be responsible for further organ hypoperfusion, independently of systemic blood flow or alteration of oxygen extraction. This is why the intensivist should urgently initiate therapies aimed at avoiding prolonged hypotension. In the case of septic shock, it has been clearly demonstrated that the area under MAP 65 mmHg was the best predictor of 30 day-mortality[38] suggesting that not only the deepness but also the duration of hypotension are important to take into account. The MAP is the driving pressure for organ perfusion when the downstream venous pressure is low. However, if the downstream venous pressure is not negligible due to organ venous congestion, the organ perfusion driving pressure is much better assessed by the difference between MAP and CVP than by the sole MAP. In other words, in case of high CVP, the CVP value should be taken into account for assessing the actual organ perfusion pressure. This could occur in some septic shock states associated with right ventricular failure or when excessive fluid resuscitation had resulted to a high CVP. When the MAP-CVP difference is considered to be too low, the clinician should consider either to decrease CVP (probably the best option when feasible) or to increase MAP or both.
To select the most appropriate treatment for increasing MAP, it is then essential to pay attention to the DAP. A low DAP is generally due to a low arterial tone and thus should prompt a vasopressive therapy urgently. As already mentioned, a low heart rate also can be responsible for lowering DAP. As a direct consequence, a low DAP in presence of tachycardia suggests that the arterial tone is totally depressed and that vasopressors should be administered in extreme urgency. When a vasopressor is administered (mainly norepinephrine), the MAP target should be individualized.[6,39] In the context of septic shock[8] or cardiogenic shock,[40] the initial MAP target should be at least 65 mmHg. The MAP target should be higher in case of history of chronic hypertension,[6,8,9,39] increased CVP or increased intra-abdominal pressure.
If the DAP is not judged as low, the low MAP is likely to be associated with an insufficient (CO) (see below). The presence of a low PP could be an additional indication of low stroke volume, although a low stroke volume can be associated with normal PP in patients having stiff arteries.
(2) Check ScvO2, a marker of the DO2/VO2 balance
In patients with shock, ScvO2 can be low (< 70%), normal (between 70 and 80%) or high (> 80%) depending on the value of DO2 and the degree of alteration of oxygen extraction capabilities.
When ScvO2 is low, as DO2 is inadequate to VO2, it is logical to attempt to increase DO2, especially in such conditions of oxygen debt, where VO2 does not meet oxygen demand.
- If Hb is low, blood transfusion can be considered.
- If Hb is not low, the insufficient DO2 should be due to an insufficient CO, either related to insufficient preload or to altered cardiac contractility. Cardiac function should be rapidly assessed by transthoracic echocardiography, which helps diagnose left ventricular dysfunction using left ventricular ejection fraction (LVEF). A low LVEF (< 45%) strongly suggests that septic shock is associated with myocardial depression and in this context, administration of an inotropic drug can be considered. Right ventricular dysfunction can be suspected when the right ventricular end-diastolic area/left ventricular end-diastolic area ratio is high (> 0.6).[41] A specific treatment can be initiated after determination of the precise underlying mechanism (mainly using echocardiography). If cardiac function is unaltered and Hb not low, the low ScvO2 should be due to insufficient cardiac preload and fluid infusion could be considered in this context. In case of any doubt, dynamic predictors of fluid responsiveness may help to make the correct decision. In mechanically ventilated patients having an arterial catheter in place and if the conditions of correct applicability of PPV are met, a PPV value > 12% is highly predictive of a positive response of fluid infusion.[23,24] If PPV cannot be measured (no artery catheter) or correctly interpreted (spontaneous breathing activity, arrhythmias, low tidal volume ventilation, low lung compliance), a PLR test can be performed to predict fluid responsiveness.[27] If echocardiography is available, the response of velocity-time integral to PLR is a good predictor of fluid responsiveness. [32] In the presence of fluid responsiveness, administration of fluids should be considered. However, as for any other therapeutic intervention, the benefit/risk ratio of fluid infusion should be individually assessed. In patients with acute respiratory distress syndrome (ARDS), the risk of worsening pulmonary edema with fluid infusion is increased due to increased permeability of the lung capillary membrane. Knowledge of EVLW and PVPI, which as good markers of pulmonary edema and of capillary leakage are helpful to assess risk of fluid infusion in such circumstances.[33] Alternatively, pulmonary artery catheterization can be considered in such complex cases.[6,7] The value of the pulmonary artery occlusion pressure can then be taken for assessing the risk of pulmonary edema formation during fluid infusion, although it is neither a marker of the volume of pulmonary edema nor of the degree of capillary leakage. Nevertheless, these advanced hemodynamic monitoring systems are relatively invasive and costly and should be reserved to situations where shock is associated with ARDS or in case of insufficient response to the initial treatment of shock.[6,7] These devices are thus particularly helpful to assess the benefit/risk ratio of fluid infusion in complex situations.
It is noteworthy that in the rare cases of preload unresponsiveness despite low ScvO2, normal cardiac function and normal Hb, it would be important to reconsider the MAP target and attempt to reach a higher value.
Presence of a normal ScvO2 while tissue hypoxia persists strongly suggests that the oxygen extraction capabilities are altered. Nevertheless, it has been clearly shown that in patients with persistent shock, hyperlactatemia and ScvO2 ≥ 70%, a further increase in DO2 (with fluids) might still increase VO2 and decrease arterial blood lactate concentration.[42] Therefore, stopping resuscitation of shock states when ScvO2 reaches a value of ScvO2 70% is a nonsense. A high value PCO2 gap (> 6 mmHg) can identify situations where increasing CO can be still attempted in spite of normal ScvO2. The PCO2 gap is a marker of the adequacy of CO with the global metabolic conditions that is less affected by impairment of oxygen extraction capacities than ScvO2. The choice of the therapy aimed at increasing CO should be based on the same reasoning than that followed in cases of low ScvO2 and normal Hb. Thus, assessment of cardiac function (using echocardiography) and assessment of fluid responsiveness (using dynamic tests) are also essential in this situation of normal ScvO2 with increased PCO2 gap. In case of normal PCO2 gap, an increase in CO is unlikely to reduce tissue hypoxia and in our opinion should not be attempted. It is likely that alteration of microcirculation and cellular metabolism disorders are responsible for the severity of shock and unfortunately cannot be positively affected by conventional therapies of shock such as fluids, vasopressors and inotropes.
When ScvO2 is higher than normal in case of persistent shock, severe alteration of oxygen extraction capacities such as arterio-venous shuntings and microcirculatory disorders are likely to exist and the prognosis is poor.[43] This essentially occurs in cases of severe hyperdynamic septic shock with multiple organ failure. It is thus illusory to think that a treatment aimed at increasing DO2 will be effective to reduce oxygen debt. Until development of new therapies targeting the microcirculation or the cell function, the intensivist is helpless with the hemodynamic failure and can only hope for a beneficial effect action of anti-infectious treatments, while maintaining as far as possible other organ functions with appropriate supportive therapies.
Conclusion
Hemodynamic management of sepsis-induced circulatory failure is complex since this pathological state includes multiple cardiovascular derangements that can vary from patient to patient. To make the correct therapeutic decision, clinicians need to be helped by hemodynamic variables other than those obtained from the clinical examination. Integration of these hemodynamic variables in a logical scheme is helpful to identify the main(s) hemodynamic disorder(s) and to assess the benefit/risk ratio of each therapeutic intervention at any time of the resuscitation phase.
 XML Download
XML Download