

 PDF
PDF Citation
Citation Print
Print



Fulminant hepatic failure (FHF) is often accompanied by a myriad of neurologic complications, including seizure, brain tissue hypoxia, and brain edema with an elevation of intracranial pressure (ICP), which are associated with high morbidity and mortality.[1] Therefore, appropriate neuromonitoring is essential for early diagnosis and to minimize secondary brain injury.[2] However, patients with FHF have a high risk of coagulopathy, which limits the use of accurate, but invasive, neuromonitoring. The suggested mechanisms of an ICP crisis in patients with FHF are 1) global brain swelling due to osmotic edema or 2) brain edema due to hyperperfusion.[1] Despite differences in pathophysiological mechanisms, those two clinical conditions are difficult to differentiate without detailed information on ICP and brain oxygen usage. Jugular venous oxygen saturation (JvO2) monitoring, well known as a surrogate of global brain oxygen consumption, has been increasingly used in the neurointensive care unit for patients with a risk of brain oxygen failure.[3,4] We report a case of successful neurological recovery based on information from JvO2 monitoring in a patient with ICP crisis due to FHF.
Case Report
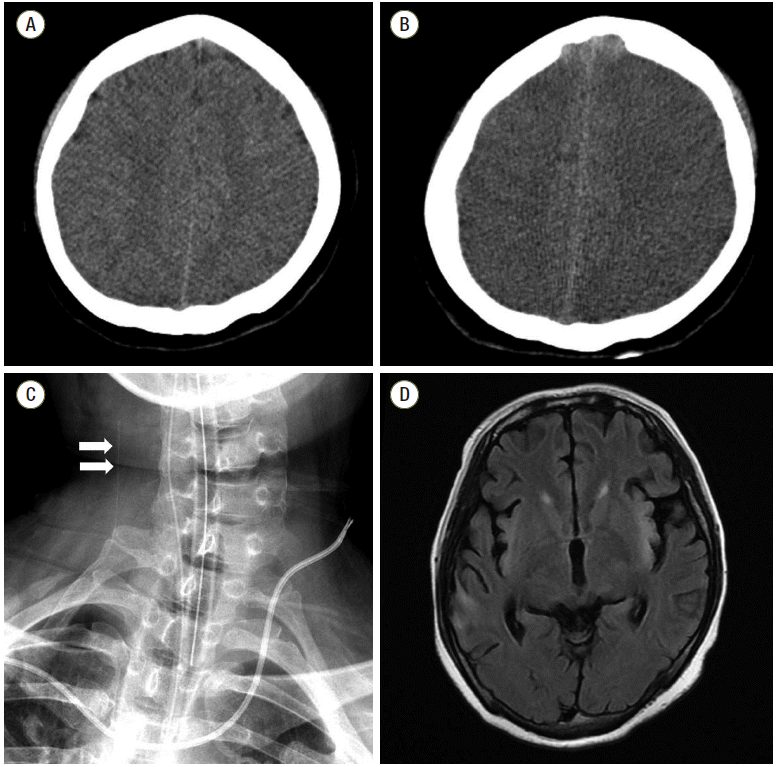
A 28-year-old woman presented to the emergency room with altered mental status. She was a heavy drinker and a carrier of the hepatitis B virus. Three days before admission, she complained of myalgia, fever, and nausea. Six hours before presentation, she became somnolent and was transferred to our hospital. On admission, she was drowsy and disoriented to time and place. Laboratory evaluation revealed elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels of 19,290 IU/L and 11,290 IU/L, respectively. Prothrombin time was 39.4 s (20%), and serum total and direct bilirubin levels were increased to 5.8 mg/dL, and 3.6 mg/dL, respectively. In addition, serum ammonia was highly elevated at 500 µg/dL and serum creatinine was 7.20 mg/dL (estimated glomerular filtration rate was 6.8 mL/min/1.73 m2). Brain computed tomography (CT) did not show any evidence of brain swelling (Fig. 1A). The patient’s condition was diagnosed as FHF with hepatic encephalopathy and continuous renal replacement therapy (flow rate 170 mL/min, dialysate 1750 mL/h, targeting for zero fluid balance) was initiated. Because the patient was young with good premorbid functional status, liver transplantation was prepared and full medical supportive care was provided.
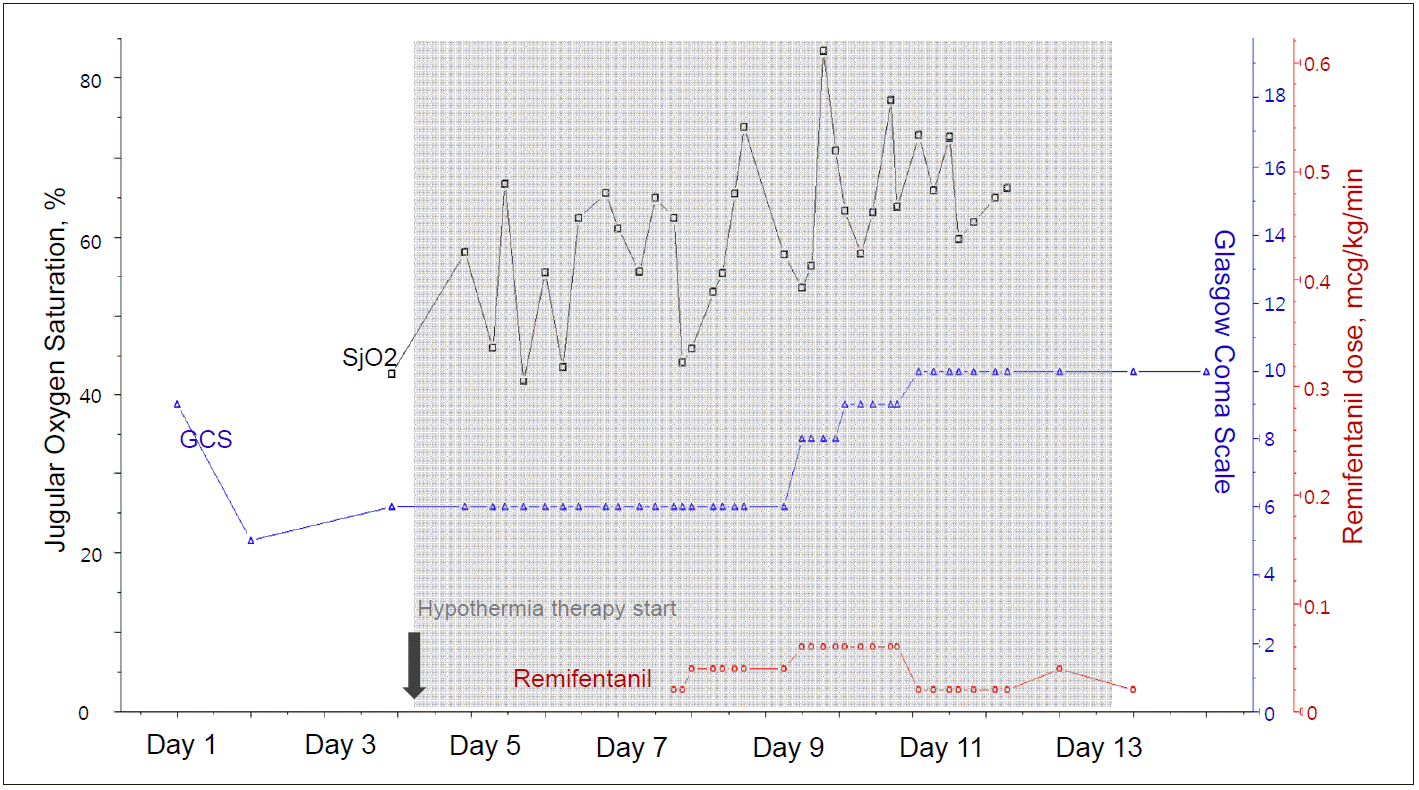
Twenty-four hours after admission, the patient suddenly became comatose (Glasgow Coma Scale [GCS] score decreased from 10 to 5) with development of generalized tonic-clonic seizures, which were controlled with intravenous lorazepam. On neurological examination, left pupillary light reflex was absent. Based on a suspicion of aggravated brain edema, treatment with 40 g of 20% mannitol was initiated, and seizures were managed with loading doses of fosphenytoin and levetiracetam. A brain CT was performed and revealed a loss of gray-white differentiation, suggestive of global brain edema (Fig. 1B). In addition to medical supportive care, 11.7% hypertonic saline (replacing mannitol) was administered to control brain swelling. Moreover, targeted temperature management was initiated at 35°C and further decreased to 33°C, to best control ICP crisis. Furthermore, to monitor global brain oxygen levels, JvO2 monitoring was initiated. The right internal jugular vein was cannulated cephalad with a pediatric central venous catheter and advanced upward (Fig. 1C). During the monitoring time, JvO2 fluctuated between 45% and 75% and did not increase above 85%, suggesting that brain swelling was mostly osmotic in origin rather than due to hyperperfusion (Fig. 2). For adequate sedation and shivering prevention, remifentanil was initiated at a rate of 0.02 µg/kg/min and subsequently adjusted based on neurological status. On day 9, pupillary light reflex was improved bilaterally and neurologic status improved from a GCS score of 6 to 8. AST and ALT levels were improved to 305 IU/L, and 1160 IU/L, respectively, and prothrombin time was 21.1 s (41%). On day 11, the patient could trace a moving object. Follow-up brain CT scan showed decreased brain swelling. Prothrombin time was improved to 14.8 s (65%). JvO2 gradually increased to 70-75% with a cessation of sedation (Fig. 2). After confirming a decrease of brain edema, a slow but controlled rewarming (0.05 °C/h) was initiated and targeted temperature management was stopped on day 12. Meanwhile, bolus doses of hyperosmotic agents were gradually reduced. On day 25, the patient could follow simple commands, such as closing their eyes or opening their mouth on request. Brain magnetic resonance image showed multiple high signal intensities in the insular cortex and basal ganglia, suggestive of sequelae of raised ICP due to hepatic failure (Fig. 1D). The patient recovered without liver transplantation.
Discussion
We report the case of a patient with FHF and concomitant ICP crisis, which was successfully treated based on the information from JvO2 monitoring.
Cerebral edema is a poor prognostic sign that renders patients at risk for subsequent tentorial herniation.[1] The literature indicates two proposed mechanisms of ICP crisis in patients with FHF: 1) cerebral edema due to osmotic swelling and 2) cerebral hyperperfusion due to the alteration of cerebral blood flow (CBF).[1] Given that the mechanisms of ICP crisis are heterogeneous in patients with FHF, a different strategy based on the causes of the elevated ICP is needed to better control the ICP surges. Invasive brain monitoring, such as ICP monitoring (Camino) or brain tissue oxygen monitoring (e.g., Licox®) may provide more accurate information, and help better differentiate these two conditions.[3-5] However, most patients with FHF have coagulopathy, which limits the use of invasive neuromonitoring. Jugular venous oxygen monitoring is less invasive compared to direct tissue brain oxygen monitoring.
JvO2 might play a role as a surrogate of global cerebral oxygen consumption. Normal oxygen extraction fraction (OEF) of the brain is approximately 30-50%. Therefore, normal values for JvO2 are 50-70%.[3,4] Based on this information, two mechanisms of ICP surges (osmotic swelling vs. hyperperfusion) can be differentiated. If the ICP surge is due to osmotic swelling, brain OEF increases due to low cerebral perfusion pressure, resulting in a reduction of JvO2 below 50%. In such cases, osmotherapies such as mannitol or hypertonic saline can be considered. On the other hand, if the ICP surge is mainly due to hyperperfusion, more oxygen than required is supplied, leading to elevation of JvO2 in excess of 80-90%. In such cases, potent vasoconstrictors such as indomethacin can be one of the treatment options.[3-6] Assuming that the patient recovered from liver failure, secondary brain damage due to ICP crisis would be the most important factor determining the neurologic outcome because her premorbid functional status was fair, except for the fact that she was a chronic hepatitis B carrier. Therefore, we chose JvO2 monitoring to facilitate the recovery. Initially the patient’s JvO2 was 55%, but it frequently decreased below 50%, requiring multiple bolus doses of hypertonic saline injections. When the patient improved on day 9, JvO2 briefly increased to 80% but rapidly improved, without use of vasoconstrictors.
Sedative agents may affect CBF by diminishing the cerebral metabolic rate and by reducing cerebral oxygen demand.[7] In our patient, CBF and JvO2 gradually increased as remifentanil dosage was reduced. However, as stated above, the accuracy of jugular venous oxygen saturation may be limited by the degree of sedation. Therefore, further research is needed to better identify factors related to JvO2 desaturation in patients with various degrees of sedation.
Moreover, in patients with hemodilution, JvO2 often drops not because of changes in brain metabolism, but owing to an increase in the OEF with low cerebral oxygen delivery. Meaningful changes were reported in severe hemodilution in a pig model (hematocrit of 7.6%).[8] In patients, however, JvO2 does not show any changes within the range of mild to moderate hemodilution (from 12 g/dL to 6 g/dL).[9] On admission, our patient’s hemoglobin level was 12.1 g/dL and was remained mostly over 10 g/dL. Therefore, we do not think that JvO2 fluctuation was simply due to a decrease in the hemoglobin level.
To summarize, we described the case of a patient with ICP crisis due to FHF. Multiple doses of osmotic agents and targeted temperature management were used to control ICP surges. Although the patient was treated under sedation, the specific cause of ICP crisis was identified based on the information from JvO2 monitoring. JvO2 monitoring is a substitute measure for global oxygen consumption and can be used for brain oxygen monitoring even in FHF patients with coagulopathy.
 XML Download
XML Download